
Clinical Researcher—September 2021 (Volume 35, Issue 7)
PEER REVIEWED
Christina Morris; Erika Stevens, MA
Pharmaceutical companies face a number of challenges, including (but not limited to) an evolving regulatory landscape; increased focus on quality metrics by governing agencies; data integrity and governance misalignment; lack of understanding customer needs; difficulty reproducing prior results for inquiries and investigations; multiple noncompliance issues; integration of quality and operations system; and a lack of process visibility (i.e., supply chain management, cycle times, bottlenecks, and shipment across sites).
Quality management system (QMS)-based principles and process automation technology are individually gaining traction on addressing some of these challenges in the pharmaceutical industry, but when combined, they solved people, process, information technology (IT), and operational issues.
Identifying the Customer(s)
QMS is a well-established systematic methodology to mitigate risk, meet compliance regulations, and improve the overall end-to-end operating model.{1} QMS methodology focuses on operational efficiency through the lens of the customer. Identifying the customer in the process or activities at hand is therefore the critical first step to understanding the “why” and “how” of the activities/process.
It is important to note that, while identifying the “customer,” the organization must be willing to look internally and/or externally, and will likely find that it has more than one type of customer. Most importantly, identifying and understanding customers and their needs and the required process controls enables a journey that is customer-centric, process focused, and co-produced with the customer. Conducting a needs assessment and determining the desired outcomes will solidify the future automation success.
Understanding the Art of Business Process and IT Strategy
After documenting a customer’s requirements and controls, the business/IT strategy should be discussed in conjunction with documenting the end-to-end value chain or process requiring analysis. To begin strategically managing the QMS (people, process, and system/data), the customer is required to rethink its practices in terms of connecting output of one activity to the input of the next activity.
For example, to complete, approve, and execute a clinical trial protocol, clinical operations staff interact with those in regulatory affairs to confirm submission of the protocol in applicable regions; pharmacovigilance staff interact with those in regulatory affairs to confirm relevant protocol safety requirements; clinical operations staff interact with those in biostatistics to confirm the statistical analysis plan; and so on.{2} Further, the activity of drafting and approving a protocol is connected to the larger process of building, conducting, managing, and closing a trial, which is connected to the even larger process of managing a product portfolio, revenue cycle, and so on.
Similar or like activities are reused/repeated multiple times by different processes in the same and/or different functions. Thus, the assessment is aimed at reviewing the following employee pain points:
- Time spent reworking work products and reports to meet specific compliance and regulatory needs.
- Misalignment of process activities within different functional quality systems documents (QSDs)/standard operating procedures (SOPs).
- Unclear link between interdependent processes and underlining IT system outputs/inputs.
- Training and how it intersects with the process, portfolio management, and commercial concept.
- Multiple technology platforms limiting process efficiency.
- Employee satisfaction and value-added tasks.
Determining the Maturity of the QMS Along the Value Chain to Meet Customer Needs
The concepts of reusability, repeatability, and sustainability are key to every QMS. To determine maturity of the QMS, the current research and development (R&D) value chain requires an assessment. An assessment is conducted on three levels to identify the maturity of process connectivity: 1) review of resources (how they are consumed in the process and systems), 2) review procedures and process (workflow and activities), and 3) review swivel chair activities in IT systems (the systems required and data input/output for the process workflow).
Upon completion of the assessment, the relative maturity of a company’s QMS is established, a customer centricity journey is documented, and the process repeatability/reproducibility of key performance indicators (KPIs) is determined. Collectively, the outputs of the assessment identify the highly manual process/activities that would substantially benefit from automation and a future state plan is reviewed and confirmed.
Designing a Fit-for-Purpose QMS
Once the level of maturity, customer’s point of view, and pain points are known and the future state agreed upon, designing a fit-for-purpose QMS begins. The basic components of a mature and well-functioning QMS include:
- People/Movement to a Quality Culture (be obsessed with failure and customer satisfaction and not perfection)
- Process/End-to-End Business Process Management (connecting the right processes together)
- Disruptive/Business Process Automation (disruptive technology and an IT framework that drives connectivity and increased production)
- Risk/Integrated Quality Risk and Control Framework (enabling holistic and offensive risk management)
- Monitoring/Quality Monitoring and Reporting (enabling real-time reporting and dashboarding)
- Training/Quality Communication and Training (connecting communication and training to everyday workflow)
- Business Continuity and Disaster Recovery (responsiveness and preemptive planning to minimize disruptions and/or outages to the research enterprise value chain)
Each of these components is essential to designing a well-connected process and data ecosystem, and must run the entire value chain—across the entire business (front office, back office, and everything in between). This connects the people through process and data across the entire value chain in a cross-functional manner—from the front office to the back office to the customer.
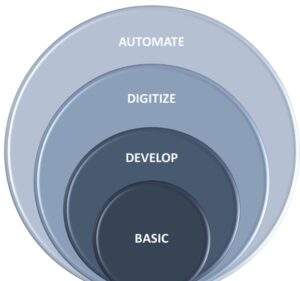
Figure 1 illustrates QMS maturity. While we could expand on all the basic components of a QMS and how to connect data in the entire value chain, this paper focuses on the rise of disruptive technologies to enable an offensive, agile, compliance-driven, and customer-focused QMS.
Figure 1: QMS Maturity
Automation and Compliance: Next-Generation Clinical Drives Offensive Future-Proofing to Improve Compliance
Compliance is a driving force for all operational procedures and activities. To build a next-generation QMS with disruptive technologies, we first examine the compliance requirements for a digital QMS within clinical trial execution. Understanding the digital compliance requirements enables us to apply the proper technology and become operationally offensive, so we started by asking the better questions:
What standards/compliance requirements are required for an integrated digital QMS in pharmaceutical companies?
To adequately address this question, a review of current requirements and regulatory guidance recommendations commenced. As 21 CFR 11 in the Code of Federal Regulations is the precedent for electronic systems and IT system audits, we began with a detailed analysis of the regulation.{3} However, 21 CFR 11 is part of a larger set of compliance standards for product/IT validation. Additionally, reviewing electronic compliance is one part of the overall review process, as there are other International Council for Harmonization/Good Clinical Practice (ICH/GCP) regulations and International Organization for Standardization (ISO) guidelines that are required to implement an integrated digital QMS.
Table 1 provides a sample review of regulations and/or standards that should be considered during the evaluation of an integrated digital QMS. Multiple guidance documents should also be considered and reviewed to confirm current regulatory recommendations.
Table 1: Sample Regulations
| Technical/IT Validation Compliance | Clinical and/or Manufacturing Compliance | Comments | |
| 21 CFR 11 | X | Gold standard for clinical IT validation | |
| ICH E6/GCP (applicable CFRs){4} | X | ||
| 21 CFR 820{5} | X | X | |
| ICH Q10{6} | X | Harmonized/Companion with ICH Q8 and Q9 | |
| ISO/IEC 9001: 2015{7} | X | Generally harmonized with ISO 13485 and 21 CFR 820 | |
| ISO/IEC 13485: 2016{8} | X | X | Generally harmonized with ISO 9001 and 21 CFR 820 |
| ISO/IEC 14971: 2019{9} | X | X | |
| ISO/IEC 17799: 2013{10} | X | ||
| REGULATION (EU) 2016/679 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL{11} | X | GDPR (General Data Protection Regulation) | |
| HITECH{12} | X | Health Information Technology for Economic and Clinical Health Act (HITECH Act) Sec. 3001; Office of the National Coordinator for Health Information Technology | |
| Cures Act{13} | X | X |
Note: CFR = Code of Federal Regulations; ICH = International Council for Harmonization; GCP = Good Clinical Practice; ISO/IEC = International Organization for Standardization/International Electrotechnical Commission; EU = European Union
Reviewing and conducting an analysis of the regulatory landscape is essential to documenting electronic requirements, user requirements, user experience, and potential user interface issues/risks for a next-generation QMS. Ultimately, the compliance analysis assists in determining (as outlined in the business case below) the disruptive digital process automation tools required to implement a process-based approach to automating the QMS.
How are the standards/compliance requirements expected to change in the next three to five years?
For more than 15 years, regulators provided industry guidance on standards and compliance requirements. Table 2 illustrates a sample of regulatory documents. Based on a review of draft U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidance documents, the focus of the major regulatory bodies will continue to be on modernizing clinical trial design, integrating clinical studies data to and from the healthcare delivery model, and addressing the risks of study quality.{14}
It is expected that the regulatory agencies will continue to create inter-agency efficiencies to enable consistent review/approval, provide consistent standards, and focus on issues critical to quality data. Regulatory standards and compliance requirements will change to reflect the need for better use of automation technology. The ability to move and analyze data in a more efficient/ effective manner will enable a holistic, risk-based approach with near real-time review, reporting, and integration of real-world evidence into clinical trials though the patient journey.
Table 2: Sample Regulations
| FDA/ICH Draft Guidance/ Consideration Documents | Investigational Product (Compound/ Molecule) |
Medical Device | Manufacturing/Distribution |
| Institutional Review Board (IRB) Written Procedures – Draft Guidance for Institutions and IRBs 08/2016{15} | X | X | |
| Acceptance of Medical Device Clinical Data from Studies Conducted Outside the United States 04/2015{16} | X | ||
| Investigational New Drug Applications (INDs) – Determining Whether Human Research Studies Can be Conducted Without an IND 09/2015{17} | X | ||
| Informed Consent Information Sheet 07/2014{18} | X | X | |
| Humanitarian Device Exemption (HDE): Questions and Answers 03/2014{19} | X | ||
| Charging for Investigational Drugs Under an IND—Q&A 06/2016{20} | X | ||
| Expanded Access to Investigational Drugs for Treatment Use—Q&A 10/2016{21} | X | ||
| Enrichment Strategies for Clinical Trials to Support Approval of Human Drugs and Biological Products 4/2019{22} | X | ||
| Exculpatory Language in Informed Consent 08/2011{23} | X | X | |
| Commercially Distributed In Vitro Diagnostic Products Labeled for Research Use Only or Investigational Use Only: Frequently Asked Questions 11/2013{24} | X | X | |
| E6(R2) Good Clinical Practice 4/2018{25} | X | X | |
| E18 Genomic Sampling and Management of Genomic Data 6/2016{26} | X | ||
| E17 General Principles for Planning and Design of Multi-Regional Clinical Trials 7/2018{27} | X | ||
| E11(R1) Addendum: Clinical Investigation of Medicinal Products in the Pediatric Population 4/2018{28} | X | X | |
| E9(R1) Statistical Principles for Clinical Trials: Addendum: Estimates and Sensitivity Analysis in Clinical Trials 5/2021{29} | X | X | |
| E2B(R3) Electronic Transmission of Individual Case Safety Reports Implementation Guide—Data Elements and Message Specification; and Appendix to the Implementation Guide—Backwards and Forwards Compatibility 2/2014{30} | X | X | |
| Q11 Development and Manufacture of Drug Substances—Questions and Answers (regarding the selection and justification of starting materials) 2/2017{31} | X | ||
| Submitting Marketing Applications According to the ICH/CTD Format: General Considerations 09/2001{32} | X | X | |
| M8 Electronic Common Technical Document (eCTD) v4.0 DRAFT Implementation Guide v2.0; and eCTD Implementation Package DRAFT Specification for Submission Formats v2.0 4/2015{33} | X | X | |
| ICH E8(R1): Revision of General Considerations for Clinical Trials 14 November 2017{34} | X | X | |
| Planning for the Effects of High Absenteeism to Ensure Availability of Medically Necessary Drug Products{35} | X |
Case Study on a Next-Generation Quality Management System
Overview
The case study focuses on an integrated and holistic approach for transforming static (written on paper) policies and procedures into process-based automation and document creation, as well as identifying and implementing automation technologies to connect and mature the QMS. The process-centric systems approach enabled identification of process improvement, attention paid to business alignment (reducing the silo work and data), and evaluation of several technologies leading to a less complex and more user-friendly, process-driven, and compliant framework for the end-user.
Process Documentation
When transforming the document management portion of the QMS, identifying and engaging with related roles, responsibilities, and guidance documents enables a culture of quality to mature. After identifying the quality culture enablers, the current functional processes are evaluated and recorded. This approach will solidify both the top-down and bottom-up change management and increase the sustainability of end-user and stakeholder adoption.
Once process analysis is completed, an inventory of relevant quality documents is mapped within the global and local process. Figures 2 and 3 illustrate the pre-automation and process landscape.
Figure 2: Pre-Automation
| Pre-Automation Process Input | Pre-Automation Process Output |
| • 65 systems
• 3,000+ datapoints/fields • 23 manual level 2 process steps with multiple level 3 and 4 activities/tasks below |
• 65 document types managed manually (which could product hundreds more, depending on the type of study)
• Duplicative data entry up to 68% |
Figure 3: Process Landscape (image intended only to illustrate complexity)
Sustainable Content and Life Cycle Transformation
Once the dependencies and other required document(s) for the process were inventoried and mapped, the following steps were taken to transform document management within the QMS:
- Prepare a new template for a process-oriented and role-focused quality management document
- Re-write quality management documents with the mature quality principles
- Review new process with stakeholders and aligned parallel processes
- Link training requirements to processes and connect with training system
- Identify and implement automation tools in an agile environment
- Explore additional technologies to enable better process and content management
Assessing and Deploying Digital Technologies
As the program was built on process, it was essential to assess the process focused on digital technologies. Based on the compliance review, user requirements were drafted and a market scan was conducted to identify applicable tool(s). Identification of a proof-of-concept (POC) process was confirmed and the POC was implemented within the system. A detailed implementation roadmap was created to support continuous and effective cross-functional process automation leveraging data reuse and data critical paths. As illustrated in Figure 4, the process of implementation is incremental.
Figure 4: Incremental Implementation
Results
The intent and scope of the business-led project resulted in an integrated, process-centric QMS document landscape. The business directives included achievement of a system that was simple, user-friendly, process-centric, and compliant. As a result of developing and delivering on the project, the return on investment (ROI) was intended to be more qualitative than quantitative. Post-automation processes are illustrated in Figure 5.
Figure 5: Post-Automation
| Post-Automation Process Input | Post-Automation Process Output |
| • 33 systems
• ~ 1,500 datapoints/fields • 23 level 2 steps |
• System produced 27 essential document types and stored documents in appropriate storage via automation
• Reduction of data entry by 48% • 6 automated level 1 processes • Realtime dashboards
|
Ongoing feedback indicated that the new process-centric document landscape was significantly easier to use. The end-user could more readily navigate the processes and find the associated documents and/or operating procedures to execute, saving them administrative time and adding real value to their task.
Additionally, compliance qualitative measures were met, as the new QMS documents and change controls passed inspection. Upon meeting the qualitative measures, the team conducted an analysis to identify qualitative ROI. Figure 6 highlights the level of impact gained through process automation. The benefits of this case study were realized over a period of 18 months and are as follows:
- The impact was greatest at the local country level, where the reduction of document maintenance was greater than 50%.
- In some cases, and in some functions, more than 80% greater efficiencies in document management were gained.
- The value and impact to the business and the end-user are substantial for both global and local levels of the organization.
- The application of QMS automation enables the same business impact, regardless of customer along the value chain.
- Further, it is expected that the increase in ROI will continue (possibly double) once automation is fully deployed.
Figure 6: Impact Level
| Global Level Impact | Local Country Level Impact | Enabling Growth Impact | |||
| 33% | 25% + | 50% + | 49% | 30% + | 25% + |
| Reduction in QMS policies and procedures document management | Reduction in new policy or procedure approval | Reduction in local policy and procedures management | Fewer local policies and procedures | Acceleration of change programs due to analysis of value chain | Acceleration in integrations and integration activities |
Conclusion
Table 3 illustrates the value created through QMS implementation. We can create critical process paths that align with critical data paths. Once these two paths converge, data are further “connected” to overall value chain processes, including front- and back-end operation activities. These principles were leveraged to merged best-in-class QMS principles with an automation platform to drive process-centric value and business efficiencies, enabling a higher level of customer satisfaction and increased risk management. The results optimized business operations and increased data throughput (reuse not reentry), removing the mundane non-value-added tasks and focusing on the pain points in the process, all the while creating a path that allows pharmaceutical companies to meet quality and compliance standards.
| Table 3: QMS Implementation Overview | ||
| Initial Client Process Assessment | QMS Implementation Goals | Value Created |
| Process documentation written to reflect topics instead of processes |
|
|
| End-users created workaround for conducting activities as finding documents was time-consuming and topic-focused |
|
|
| Lack of clarity on which process documents or sections apply to a specific role |
|
|
| Training plans did not support compliance within the process (input and output, reference to relevant prior or subsequent tasks, overall enterprise value chain training, process to enterprise requirements) |
|
|
References
- https://www.sciencedirect.com/science/article/pii/S1877042812045958
- Morris C, Stevens E. 2021. Fixing the foundation: Using digital strategy and process automation in a disruptive approach to R&D optimization in the life sciences. Clinical Researcher 35(3). https://acrpnet.org/clinical-researcher-april-2021/
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/part-11-electronic-records-electronic-signatures-scope-and-application
- https://www.fda.gov/files/drugs/published/E6%28R2%29-Good-Clinical-Practice–Integrated-Addendum-to-ICH-E6%28R1%29.pdf
- https://www.fda.gov/medical-devices/postmarket-requirements-devices/quality-system-qs-regulationmedical-device-good-manufacturing-practices
- https://www.fda.gov/media/71553/download
- https://www.iso.org/standard/62085.html
- https://www.iso.org/standard/59752.html
- https://www.iso.org/obp/ui#iso:std:iso:14971:ed-3:v1:en
- https://www.iso.org/standard/39612.html
- https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016R0679
- https://www.hhs.gov/sites/default/files/ocr/privacy/hipaa/understanding/coveredentities/hitechact.pdf
- https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act
- https://database.ich.org/sites/default/files/E6-R3_FinalBusinessPlan_2019_1117.pdf
- https://www.federalregister.gov/documents/2016/08/02/2016-18191/institutional-review-board-written-procedures-guidance-for-institutions-and-institutional-review
- https://www.fdanews.com/ext/resources/files/04-15/04-27-15-trials.pdf?1483870630
- https://www.fda.gov/files/drugs/published/Investigational-New-Drug-Applications-%28INDs%29-Determining-Whether-Human-Research-Studies-Can-Be-Conducted-Without-an-IND.pdf
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/informed-consent
- https://www.federalregister.gov/documents/2014/03/18/2014-05900/humanitarian-device-exemption-questions-and-answers-draft-guidance-for-humanitarian-device-exemption
- https://www.federalregister.gov/documents/2016/06/03/2016-13166/charging-for-investigational-drugs-under-an-investigational-new-drug-application-questions-and
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/expanded-access-investigational-drugs-treatment-use-questions-and-answers
- https://www.fda.gov/media/121320/download
- https://www.fda.gov/files/about%20fda/published/Exculpatory-Language-in-Informed-Consent—Draft-Guidance.pdf
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/distribution-vitro-diagnostic-products-labeled-research-use-only-or-investigational-use-only
- https://www.fda.gov/files/drugs/published/E6%28R2%29-Good-Clinical-Practice–Integrated-Addendum-to-ICH-E6%28R1%29.pdf
- https://www.ema.europa.eu/en/ich-e18-guideline-genomic-sampling-management-genomic-data
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e17-general-principles-planning-and-design-multi-regional-clinical-trials
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e11r1-addendum-clinical-investigation-medicinal-products-pediatric-population
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e9r1-statistical-principles-clinical-trials-addendum-estimands-and-sensitivity-analysis-clinical
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/e2br3-electronic-transmission-individual-case-safety-reports-implementation-guide-data-elements-and
- https://www.federalregister.gov/documents/2017/02/21/2017-03309/q11-development-and-manufacture-of-drug-substances-questions-and-answers-regarding-the-selection-and
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submitting-marketing-applications-according-ichctd-format-general-considerations
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m8-electronic-common-technical-document-ectd-v40-draft-implementation-guide-v20-and-ectd
- https://database.ich.org/sites/default/files/E8-R1_EWG_Concept_Paper_0.pdf
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/planning-effects-high-absenteeism-ensure-availability-medically-necessary-drug-products-0

Christina Morris is Vice President for Digital Transformation with Hoverstate, and has worked as a Clinical Research Associate and in clinical operations, quality assurance, and process improvements for a variety of organizations. She has been designing and implementing digital process automation applications across the life science value chain for more than eight years.

Erika Stevens, MA, is the 2021 Chair of the Association Board of Trustees for ACRP and leads Transformation Advisory Solutions for Recherche Transformation Rapide.


