
Clinical Researcher—February 2022 (Volume 36, Issue 1)
PEER REVIEWED
Dr. Kumari Priyanka, BDS, PGDCR; Tejas Thomas, MSc, PGDCR; Pullareddy Medikonda, MPharm; Dr. Shubha Rao, BDS, MS; Vladimir Penkrat, MBA
Since 2014, clinical trials within the European Union (EU) are conducted in accordance with the established EU Clinical Trials Regulation (CTR). The European Medicines Agency (EMA), in collaboration with the European Commission and EU Member States, is developing a centralized portal and database, the Clinical Trials Information System, to harmonize all clinical trial requirements and improve trial data transparency across the EU. This paper will briefly focus on the impact of the transition from the existing processes and systems, including the European Union Drug Regulating Authorities Clinical Trials (EudraCT) database, to the forthcoming centralized system. Furthermore, it will focus on a perception on the ways to overcome the potential challenges that may be experienced by study sponsors during this transition.
| CRO | Contract research organization |
| CTA | Clinical trial application |
| CTD | Clinical Trials Directive |
| CTIS | Clinical Trials Information System |
| CTR | Clinical Trials Regulation |
| EC | Ethics committee |
| EEA | European Economic Area |
| EMA | European Medicines Agency |
| EOT | End of trial |
| EU | European Union |
| EudraCT | European Union Drug Regulating Authorities Clinical Trials |
| GCP | Good Clinical Practice |
| IT | Information technology |
| MS | Member States |
| RFI | Request for information |
Background
The conduct of any clinical trial generates extensive patient data, which are very sensitive in nature. Revealing any such data requires a unique approach in presenting the key trial data to regulatory authorities, healthcare professionals, patients, or other sponsors. Currently, clinical trial conduct in the EU is highly regulated and requires trial registration with the EudraCT database.{1,2}
In April 2014, the European Parliament and the EU Council released the Clinical Trial Regulation (EU-CTR) No 536/2014 regarding clinical trials on medicinal products for human use repealing EU Clinical Trial Directive (EU-CTD) 2001/20/EC. The primary goal of this new regulation is to protect the rights, safety, dignity, and well-being of the trial participants and to generate reliable and robust clinical trial data. The regulation was intended to facilitate harmonization of the assessment on medicinal products and supervision of processes for conducting clinical trials throughout the EU using a centralized Clinical Trials Information System.{3,4} The highlights of EU-CTD 2001/20/EC and EU-CTR 536/2014 are presented in Figure 1.
Figure 1: Comparison of 2001 Directive with the New 2014 EU-CTR

(CT=clinical trial); Source: CTIS-highlights, December 2020.{5}
The 2014 regulation will be fully implemented after the publication of the notice confirming the full functionality of the EU portal and the EU database by the European Commission.{4}
CTIS—A Cornerstone of the Clinical Trials Regulation EU No 536/2014
The key highlight of EU-CTR 536/2014 is to provide a single, unified portal and database, which is the Clinical Trials Information System (CTIS), available for both trial sponsors and regulatory authorities of each Member State. The CTIS will be a centralized, paperless, integrated, single-entry point for submission, evaluation of data, authorizing, supervising, and reporting any trial-related information between the relevant Member States. The EMA will set up and manage the CTIS, in collaboration with the Member States and the European Commission.{5} The purpose of this system is to considerably facilitate the process of clinical trial conduct across EU, starting from the initial submission to authorization, providing corrective measures, inspection information, and publication of relevant documents for the general public and substantial updates over time.{3,4}
Overview of CTIS Workspaces
The CTIS will provide dedicated workspaces to enable sponsors, regulatory authorities, and the public to access a suite of functionality. For sponsors and regulatory authorities, common roles in the portal include User Administration, Clinical Trial Information, Notices and Alerts, and Annual Safety Reporting. The various workspaces within CTIS include:
- Sponsor Workspace
Sponsors, academics, and others (including regulatory project managers, in-country specialists, and CTIS submission managers) will use the Sponsor Workspace on the tool, enabling them to apply for their trial authorization using a single application for up to 30 EU/European Economic Area (EEA) countries.
- Authority Workspace
Member States from different participating countries, national competent authorities, and ECs can coordinate and promote harmonization of clinical trial assessment decisions and administrative processes.
- Public Portal
The general public will be able to access clinical trial data in all official EU languages in compliance with the disclosure policies (EU-CTR 536/2014 and Policy 0070). Data and information submitted through the EU portal will be stored in the EU database, and shall be publicly accessible unless confidentiality is established on any grounds as stated in Article 81, paragraph 4 of the EU-CTR 536/2014.
A pictorial representation on the functions of CTIS for the different user workspaces is provided in Figure 2.
Figure 2: Functions of CTIS for User Workspaces
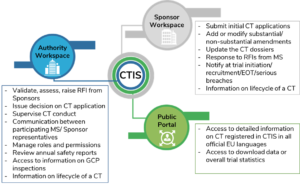
(CT=clinical trial); Source: CTIS-highlights, December 2020.{5}
Dedicated workspaces with restricted access to multiple stakeholders are expected to enhance support in daily business processes throughout the lifecycle of a clinical trial. Business experts are focused on continuous development of the CTIS to ensure whether it is fit for purpose. Since June 2019, the areas of consideration included document management, enhanced submission, and facilitating overall scientific and regulatory review and monitoring.{6} In April 2021, following a successful independent audit, the EMA’s Management Board confirmed that CTIS is fully functional and meets the specifications as stated in the Article 82, Paragraph 2 of the EU-CTR. The European Commission will further publish a notice in the Official Journal of the EU, six months before the planned CTIS launch date.
Benefits of the CTIS Workspaces
The CTIS is intended to provide an improved and more collaborative environment for the user community. Some of the key benefits of the system include efficiency, increased transparency, patient protection, and better capability to facilitate effective submission processes (see Figure 3).
Figure 3: Key Benefits of the CTIS
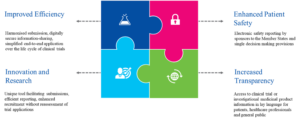
Source: CTIS-highlights, December 2020.{5}
Transition to the New Regulatory System
The new clinical trial system is planned to be fully functional after a three-year transition period (2022–2025) for all clinical trials to go through CTIS.{7} The EMA announced the CTIS go-live date as January 31, 2022. During the first year, new clinical trial applications (CTAs) may be submitted either under the new regulation (via the EU portal and database) or under Directive 2001/20/EC. Over the second and third years (from January 31, 2023 onward), all initial CTAs will be submitted through CTIS; however, old trials will continue to be governed by the Directive. From January 31, 2025 onward, all ongoing clinical trials will be governed by the Regulation and must go through CTIS.{8}
A well-planned CTIS training programme is already under way to provide users with the required skills, capabilities, and knowledge for successful adoption of CTIS.{5} EMA’s training materials are tailored for clinical trial sponsors and staff of the EU Member States, European Commission, and other organizations who will use the system. Presentations, quick guides, frequently asked question sheets, e-learnings, webinars, and short videos are available online, which will help in the preparedness for CTIS end-users. The training is scheduled in three main programme streams for sponsors:
- Sponsor Master Trainers are conceptualized to use the train-the-trainer approach for the Member State users aimed at CTIS functions applicable for commercial sponsors and contract research organizations (CROs), which are likely to submit several CTAs and have multiple CTIS users across various organization models.
- A two-day webinar for users from small and medium-sized enterprises, academia, and other non-commercial clinical trial sponsors will disseminate the knowledge on applicable CTIS functions.
- Training catered on specific CTIS functions was planned to be initiated in the fourth quarter of 2021 for end-users from all sponsor groups.{9}
More information on the CTIS training programme is available here.
An overview and a high-level illustration on the possible lifecycle of a clinical trial via the new portal, supporting various user groups including sponsors, Member States, the European Commission, and the public is presented in Figure 4.{10}
Figure 4: Lifecycle of Clinical Trials in CTIS
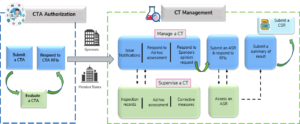
(ASR=annual safety report, CT=clinical trial, CSR=clinical study report); Source: Guide to CTIS Training Material Catalogue.{10}
Potential Challenges for Sponsors During the CTIS Transition
Currently, separate CTAs and submissions are globally initiated and updated locally by the in-country personnel for multiple Member States. With the introduction of CTIS, sponsors need to assess change in the processes across the lifecycle of a clinical trial. Most of the users from sponsors, small and medium-sized enterprises, and academia are now involved in allocating time and resources to align with the transition to the new system. Many users with limited resources are also dependent on CROs, making it a huge responsibility to decide on the user management for each trial, especially for those complex trials with more than one vendor.
Nevertheless, the transition to the new system is leading to increased risks regarding trust, accuracy, costs, and workload. While the small and medium-sized enterprises are planning to train on the available resources, the large sponsors are preparing to align several hundreds or thousands of resources with the new processes.{11} Some of the possible common challenges faced by sponsors are listed below:
1. Administrative burden for new user access: Sponsors will need to configure Administrative User accounts, which will be obtained only upon EMA’s validation of certain documents, to perform their responsibilities and meet their business needs. Administrative sponsors will be responsible for user management by assigning, amending, revoking, or approving user roles; therefore, the more users there are tied to a sponsor, the more responsibility there will be for the sponsor administrator. It is therefore critical for sponsors to maintain the right balance in assigning roles to their resources within the CTIS.
2. Training for end-users: Sponsors need to carefully identify the resources (or vendors) who will undergo the EMA training programme and in turn develop training materials and train other resources within the organization. The programme should be lined up with the roles assigned in CTIS to ensure that employees efficiently perform their activities within the stipulated timelines.
3. Revealing sensitive data: As CTIS has open access for the general public, it is critical for the sponsors and/or any end-users to ensure that no personal or sensitive information is used within the CTIS while considering the transparency guidelines wherever appropriate. Coordinating with translations and redaction of trial-related documents in multiple languages could possibly burden the trial timelines.
4. Increased cost and administrative burdens: Sponsors and institutions were accustomed to their company-specific standard operating procedures and workflows in preparing submission documents as per country-specific requirements, which may need to be revised to cater to the new requirements. In addition, essential trainings for individuals and upgrades in the IT infrastructure could lead to increased costs and administrative burdens.
5. Tight submission timelines: Upon the implementation of CTIS, sponsors need to closely coordinate with regulators for initial applications and all subsequent clinical trial modifications. Any lapse in the timeline might result in legal consequences, such as lapsed application; therefore, sponsors need a strong strategic plan and timelines defined for all studies of the same product. Ongoing assessment in one Member State will put a hold on any substantial modifications for other Member States.
Other Considerations for Preparedness for CTIS
Planning new clinical trials: Sponsors planning to conduct any new trials within the EU should be mindful of the transition period and ensure compliance either with the old Directive or new Regulation, based on the duration of planned clinical trial. In addition, it is important to plan timelines and trial-related activities well in advance.
Adaptability to the new Administrative User system: As CTIS will have dedicated sponsor workspaces, all sponsors (including institutions, academia, or other organizations) should develop the expertise to adopt and compile CTA dossiers in compliance with regulations for any new or ongoing trials on the new secure workspace.
Well-established IT Infrastructure: Sponsors will need to upgrade their IT/technical/ infrastructure to gain access to the new secure workspace for preparing and compiling data submitted to the Member States for assessment of any new and updated trials, and to avoid any delays. Further IT infrastructure upgrades may be necessary whenever the CTIS platform is updated.
Define workflows: Sponsors may need to create new or revised standard operating procedures and workflows for dossier preparation, compilation of all CTA authorizations and related documents for multiple Member States assessments through the CTIS.
Conclusion
The much-awaited digital transformation of the healthcare industry has been setting its roots for almost a decade. The EU-CTR and CTIS will streamline the process for seeking approval for the conduct of clinical trials and harmonize the end-to-end electronic application procedures over the lifecycle of trials across the EU.
It is expected that the new, centralized CTIS will improve collaboration among sponsors, innovators, researchers, Member States, and regulatory authorities with enhanced patient safety and knowledge-sharing, reduced delays in clinical trial approvals, and increased efficiencies in trials within the EU. Furthermore, it is expected to prevent redundancy in clinical trials and unnecessary duplication of research.
The fully implemented EU-CTR will foster innovation and competitiveness of European clinical research by bringing together methodological data quality and ethical integrity. It will further enhance trial data transparency for improved patient safety.{4}
A well-planned and detailed training programme on CTIS has been initiated by the EMA, in order to prepare all the end-users ahead of its launch. The CTIS training material is also tailored in various modules for different end-users based on their roles and responsibilities. It is important that every responsible individual complete the prerequisites to fully enable a smooth transition to the new and secure system.
References
1. https://www.clinicaltrialsregister.eu/
2. https://www.efpia.eu/about-medicines/development-of-medicines/regulations-safety-supply/clinical-trials/sharing-clinical-trial-information/
3. https://ec.europa.eu/health/sites/default/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf
4. https://www.ema.europa.eu/en/human-regulatory/research-development/clinical-trials/clinical-trial-regulation
5. https://www.ema.europa.eu/en/documents/newsletter/clinical-trials-information-system-ctis-highlights-december-2020_en.pdf
6. https://www.ema.europa.eu/en/documents/report/european-medicines-agencys-interaction-industry-stakeholders-biennial-report-2018-19_en.pdf
7. https://www.ema.europa.eu/en/documents/presentation/presentation-ctis-harmonising-submission-authorisation-supervision-clinical-trials-p-vankeerberghen_en.pdf
8. https://www.ema.europa.eu/en/documents/newsletter/clinical-trials-information-system-ctis-highlights-august-2021_en.pdf
9. https://www.ema.europa.eu/en/documents/other/clinical-trials-information-system-ctis-training-information-update-progress_en.pdf
10. https://www.ema.europa.eu/en/documents/other/guide-ctis-training-material-catalogue_en.pdf
11. https://www.ema.europa.eu/en/documents/presentation/presentation-how-sponsors-are-preparing-ctis-large-sponsor-perspective-r-m-swallow_en.pdf

Dr. Kumari Priyanka (kumari.priyanka@indegene.com) is a Manager in Regulatory Solutions department with Indegene Pvt Ltd, Bangalore, India and lead author of this article.

Tejas Thomas (tejas.thomas@indegene.com) is a Team Lead in Regulatory Solutions department with Indegene Pvt Ltd, Bangalore, India.

Pullareddy Medikonda (pullareddy.medikonda@indegene.com) is an Associate Manager in Regulatory Solutions department with Indegene Pvt Ltd, Bangalore, India.

Dr. Shubha Rao (shubha.rao@indegene.com) is a Senior Director and Practice Head of the Regulatory and Safety Business Unit with Indegene Pvt Ltd, Bangalore, India.

Vladimir Penkrat (Vladimir.Penkrat@indegene.com) is an Associate Vice President – Global Head of Safety & Regulatory Affairs with Indegene Inc., Princeton, N.J.


