
Clinical Researcher—January 2019 (Volume 33, Issue 1)
PEER REVIEWED
Manfred Stapff, MD, PhD; Jennifer Stacey
Drug development is a long and complex process. A drug’s life cycle is not limited to the results of clinical trials in Phase I through Phase IV—it also includes research about the cause and natural history of diseases, clinical outcomes, long-term safety, tolerability, optimal treatment targets, and new indications.
Over the past decade, key performance metrics in the clinical trials area have been constantly disappointing, and even worsening.{1,2} As the complexity of clinical trial protocols increases, the feasibility to conduct them and the percentage of sites fulfilling the enrollment goal decreases.{3}
Clinical research, especially as applied to drug development, is a very data-driven industry. Biostatistical methods and the interpretation of study results based on p-values dominate the scientific decision-making processes. However, in planning and operating clinical research, objective data and metrics are not used to the extent to which they are available today.
For example, healthcare data have been available in electronic format in recent years. The digitalization of the U.S. healthcare system, driven by the 2014 “Meaningful Use” legislation, can be considered almost complete. By 2015, 96% of all hospitals in the United States had already adopted certified electronic health records.{4} However, the electronic data thus far have been locked in various systems, are scattered in many locations, and follow different standards of ontology, units, and other characteristics. These electronic data issues, together with the lack of a consumable, user-friendly visualization platform, made the use and interpretation of this vast amount of medical information difficult.
The following is intended to provide an overview of how electronic health data can currently provision the design and conduct of clinical trials, as well as support other medical research areas.
Electronic Medical Records Versus Claims Data
The two major sources for real-world data (RWD) in medical research are electronic medical records (EMRs) and insurance claims data. EMRs can be considered as the more “medical” component of patients’ health information. They contain data about diagnoses, examinations, and treatments as documented by a provider who applies healthcare to a patient.
A patient’s EMR data from a specific healthcare provider may be blind to information collected by other providers. For instance, another hospital visited (or physician consulted) by a patient may not have access to the patient’s prior EMR information, as these records may be in separate repositories unique to each treating healthcare organization.
Meanwhile, claims data represent the more “administrative” part of a patient’s health history. They originate from the interaction between provider and payer, and could include documentation which have been submitted or adjudicated or remitted for payment. Because of the original billing related intention, claims data may be limited to information supporting reimbursement (see Table 1).
Table 1: Claims Data Compared to EMR Data
| Claims Data | EMR Data | |
| Scope of data | Broad: Information from all doctors/providers caring for a patient | Limited: Only the portion of care provided by doctors using the specific EMR of a provider organization |
| Contained diagnoses | Limited to diagnoses supporting a claim | Complete set of conditions and comorbidities |
| Included patients | Payers’ covered population, U.S.-employed socioeconomic group | All patients of a healthcare provider, including uninsured |
| Medication | All prescriptions that were filled, including dates of refills | Knows only that a physician prescribed at drug, but not if it was filled |
| Longitudinality of data | Payer/employer-based: As long as a patient stays with the same insurance | Provider-based: As long as the patient stays with the same healthcare provider |
| Richness of data | As necessary for reimbursement (diagnoses, procedures, treatments) | More complete medical picture (diagnoses, laboratory results, vital signs, problem list, etc.) |
| Timeliness | Lag time, delay from submit to close | Often real time, as soon as entered/coded |
As a rule of thumb, one can assume that claims data better support studies about the economic effect of a therapy or the cost burden of a disease, while EMR data better capture natural disease history, efficacy, safety of a drug, or the outcome of a disease. Ideally, both data sources, used together on a patient level (also known as “linked data”) and cleaned for duplicate information, would provide an optimal dataset for all applications.
Incorporating additional data sources can enhance patients’ health histories and clinical characteristics beyond standard medical coding practices. For example, data from tumor registries often contain tumor stage at diagnosis, histology, and other cancer-specific factors; a genomics database may include details on sample sites tested and variant types. Such sources can open the world of personalized medicine in a data context.
How Can RWD Support Clinical Trials?
The success rate of drug candidates making it all the way from Phase I to launch remains low (approximately at 10%).{5} Meanwhile, the complexity of clinical trial protocols, notably expressed by number of patient eligibility criteria requirements, is increasing, and this leads to significant enrollment challenges.{6} Less than 30% of protocols do not need to be amended; 70% need two to three changes over the course of the study, which is an inefficient trend causing damages in terms of costs and time.
Study results are only taken seriously if there is a p-value below 0.05 or an appropriate confidence interval. However, in conducting clinical research, data and analytics are not used to the extent they are available.
When determining the target population for clinical trial protocol design—from eligibility criteria to whether an amendment would improve a study—protocol authors traditionally rely on literature, experience, or expert opinion. They often do not have access to or use the extensive amount of RWD available in ways which would propel this decision-making process into a more objective and real-world scenario. Many costly and time-consuming amendments could be avoided by proper data-driven strategy planning.{7}
Where in the Clinical Trial Process Can We Use RWD?
Trial design: Protocol authors need reliable and real-world information about patients, diseases, comorbidities, and concomitant treatments from routine medical practice (i.e., how patients present themselves in a true medical setting). RWD allow these authors to design clinical trial protocols in a realistic manner, including creating a feasible set of eligibility criteria.
A very simple example: It is a well-known fact that elderly patients and minorities are usually underrepresented in clinical trials.{8} Upper age limits are introduced in trial protocols for safety reasons, but often shift the demographics of the population toward younger patients. A quick look at RWD and the age distribution of patients with the target indication helps to quantify the discrepancy, and to set the age limit to an optimal value with the best balance between safety and representativeness (see Figure 1).
Figure 1: RWD Age and Gender Distribution of Patients with Rheumatoid Arthritis (RA) on Disease-Modifying Therapy
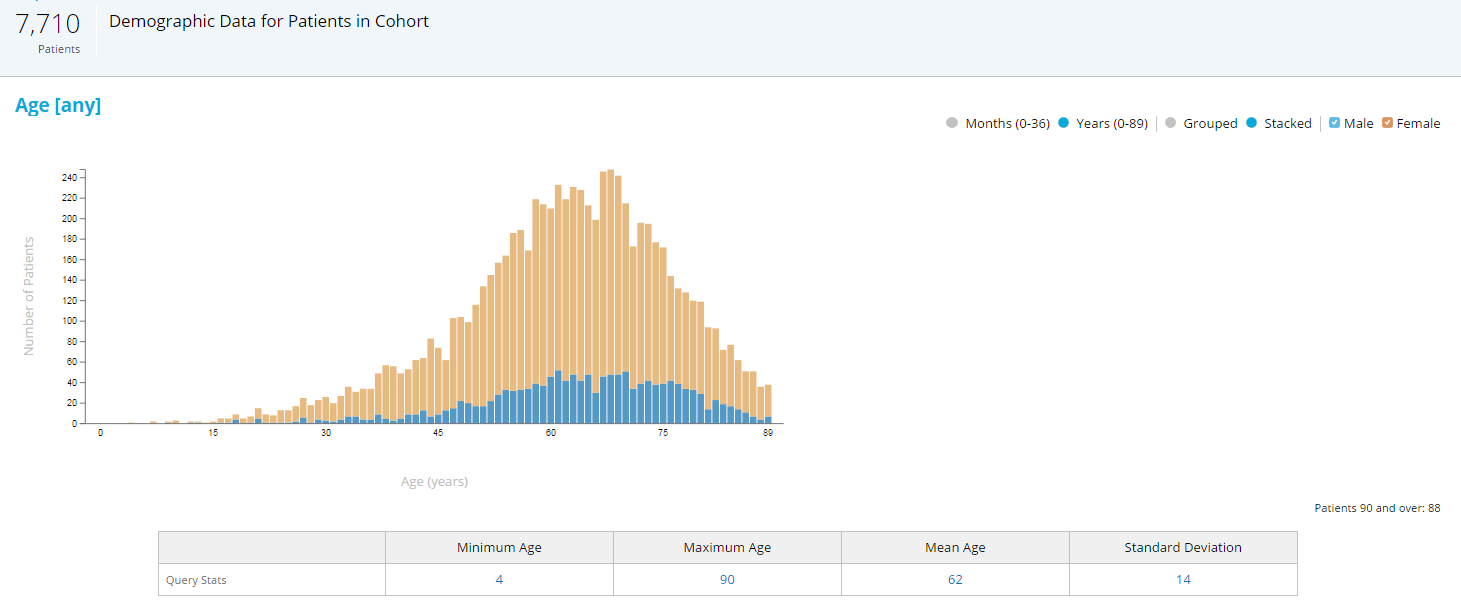
Note: If the upper age limit was set to 75 years versus 90 years, the study would miss almost 20% of patients with RA.
Study Feasibility: Clinical trial eligibility criteria are often compiled arbitrarily and carried forward through development phases by company standards (“tradition”), or come from individual(s) expert input(s). Nevertheless, they are rarely tested against RWD, especially in terms of their effects on the final percentage of eligible patients with all criteria taken into consideration together.
A simulation of a criteria analysis, or “patient funnel,” can help identify the most impactful criteria, predict recruitment hurdles, and test the effect on enrollment if criteria are changed (see Figure 2).
Figure 2: Automated “Patient Funnel” to Simulate Enrollment and Effect of Eligibility Criteria on Patent Availability (Based on RWD)
Note: Patients with cardiovascular events, with controlled hyperlipidemia, receiving statins, and with different cholesterol lab values were compared.
Site Selection: Once the protocol is designed, the study should be placed only in those sites where there is proof of availability of eligible patients. Traditionally, lengthy and time-consuming feasibility questionnaires are used to determine the number of potentially eligible patients at a site, and often this is estimated by an investigator. An RWD system which keeps the link from anonymized patient data back to the site—ideally with a built-in communication feature to the sites—allows the user to select sites with pre-screened patients. It also simultaneously addresses the Good Clinical Practice requirement{9} of the investigator to prove access to suitable study subjects.
Patient Screening: Data privacy regulations require that the collection of patients’ health data happens in an anonymized, or at least pseudonymized, manner that makes re-identification of individuals impossible. A federated network structure, however, allows aggregated statistical counts to be obtained from the data source, keeping the original data at the source together with an identification key. Therefore, this enables the site via an “Honest Broker” to re-identify eligible patients (those matching inclusion/exclusion criteria) and potentially contact them (after respective institutional review board approval) for study participation.
Many vendors and data providers offer service or systems for different aspects of clinical trial optimization. The more steps of the process, from protocol design to patient enrollment, that can be addressed by the same system, the easier it will be for the corporate sponsor or the contract research organization it is using to implement such support from the procurement, budgeting, efficiency, and training perspectives.
From Randomized Controlled Trials to Real-World Evidence Studies
In decades past, randomized controlled trials (RCTs) represented the one and only gold standard for gaining scientific knowledge in drug development and in medicine in general.{10} Only with the advent of EMRs has a new method, generally referred to as real-world evidence (RWE) studies, been discussed as reasonable and more representative alternative to RCTs.
RCTs are usually conducted in a very experimental and unrealistic setting. Nowhere in actual medical practice are patients so carefully selected, so closely supervised, and so well cared for as they are in a clinical trial. In some therapeutic areas, such as oncology, the expectations of study subjects achieving efficacy are quite high, and clinical trials are intensively promoted as best treatment options. Thus, a significant placebo effect can occur, and ethical questions are often raised.{11}
Inclusion of patients who may be more likely to show efficacy and exclusion of patients with certain risk factors dramatically reduce the representativity of the study cohorts and their applicability to the general population.
On the other hand, RWE studies have their flaws, too. The main subject of criticism concerns the quality and completeness of the data, especially in terms of the trustworthiness of data analyses when there has been no randomization of the subjects. In comparative RWE studies, the susceptibility for confounding factors (bias) requires correcting or balancing methods, such as stratification or propensity scoring, to achieve comparable cohorts. See Table 2 for a summary of the general differences between RCTs and RWE studies.
Table 2: Main Differences Between RCTs and RWE Studies
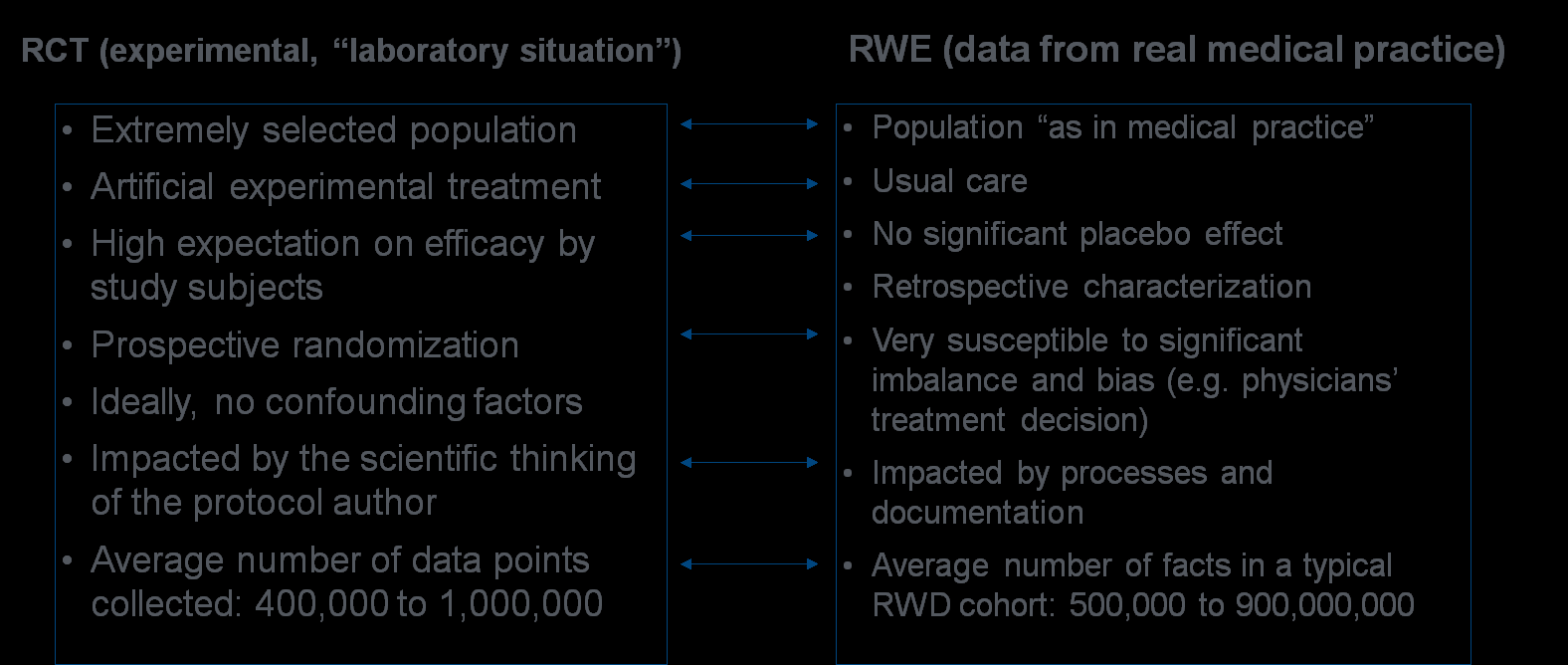
The data from an RWE study cohort can contain much more information than the data from an RCT. This is mainly due to the much larger sample size, and often longer observation period, that can easily be achieved in comparison to RCTs with limited durations in which patients are willing to participate.
In RCTs, only a fraction of the available evidence is used. Estimates report that only about 2% of patients with cancer can enroll into a clinical trial, but we use 100% of the information from this highly selected small population for decision making about that specific oncology condition.{12}
Data in RCTs are perfectly validated against the source and checked for errors, as they are heavily regulated and monitored. Contrarily, RWE studies take the data as they are, reflecting actual medical practices. As such, they are influenced by patient characteristics (demographics, comorbidities) and by provider characteristics (prescribing behavior and documentation completeness).
Meanwhile, it is sometimes the case that not enough discipline is applied in differentiating between RWD and RWE. In general, the “data” in RWD are related to the delivery or reimbursement of healthcare to a patient; they only become the “evidence” in RWE if adequate methods of collection, analysis, and interpretation of the data are applied. Only the combination of high-quality data collection and proper scientific methodology creates RWE out of RWD and makes this RWD/RWE combination “fit for purpose.”
The more accessible RWD become and the more valid RWE analyses are considered as researchers’ capabilities to do so develop and improve, the more questions will be raised over the extent to which RWE studies will one day replace RCTs. In our view, while RCTs are certainly complicated and costly, they will most likely never be replaced completely by RWE studies.
For drug development, especially in early phases when the knowledge about safety and efficacy of an experimental therapy is very limited, researchers may always need the experimental and relatively safe environment of a clinical trial. Yet in the advanced stages of clinical development—in Phase IV or perhaps even late in Phase III—RWE studies can be a much more cost-efficient tool for collecting the necessary knowledge based on a conditional approval for new indications or for long-term safety observations.
Proper Analyses Methods Needed for RWE Studies
Due to the ease of use and cost efficiency, it may be tempting to run repeated analyses on RWD until a desired result is found, and then take this result as scientifically proven. Terms like “data dredging,” “fishing expeditions,” “p-hacking,” and “selective publishing” are used to describe this undesirable practice. Therefore, it is extremely important to follow proper scientific methods from concept to planning regarding data collection, analysis, interpretation, and publication.
Ideally, an RWE platform would require a predefined analysis plan to be uploaded and documented, and would have an audit trail which date stamps all analytical steps. This would show that the pre-specified data analysis plan was followed, and no result-driven analysis was conducted.
Proper documentation of analytical steps is important for the overall credibility of the study, and for the use of RWE within the context of meeting regulatory expectations for validity of data comparable to what is seen from RCTs. Guidelines are being developed and standards are currently being defined by several organizations, including the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) and the International Society for Pharmacoepidemiology (ISPE){13}, as displayed in Figure 3.
Figure 3: ISPOR and ISPE Recommendations for Good Procedural Practices for Hypothesis Evaluating Treatment Effectiveness Studies
- A priori, determine and declare that a study is a Hypothesis Evaluation Treatment Effectiveness (HETE) study or an exploratory study based on conditions outlined below.
- Post a HETE study protocol and analysis plan on a public study registration site prior to conducting the study analysis.
- Publish HETE study results with attestation to conformance and/or deviation from the study protocol and original analysis plan. Possible publication sites include a medical journal or a publicly available website.
- Enable opportunities to replicate HETE studies (i.e., for other researchers to be able to reproduce the same findings using the same dataset and analytic approach). The ISPE companion paper lists information that should be reported to make the operational and design decisions behind an RWE study transparent enough for other researchers to reproduce the conduct of the study.
- Perform HETE studies on a different data source and population than the one used to generate the hypotheses to be tested, unless it is not feasible (e.g., another dataset is not available).
- Authors of the original study should work to publicly address methodological criticisms of their study once it is published.
- Include key stakeholders (patients, caregivers, clinicians, clinical administrators, payers, regulators, manufacturers) in designing, conducting, and disseminating HETE studies.
Studies using RWD have not yet achieved the levels of credibility and sophistication credited to RCTs, with their very detailed guidelines and regulations. While the U.S. Food and Drug Administration has already issued guidelines for the use of RWD in the regulatory process of medical devices, similar guidelines for use in drug development are pending. Therefore, RWE studies will, for the time being, mainly focus on hypothesis-generating projects, signal detection in pharmacovigilance, and obtaining supportive data for new indications (see Figure 4).
Figure 4: Potential Use Cases for RWD in Medical Research and Drug Development
Clinical Development and Operations
Use in design and statistical planning (sample size, variability, event rates)
Test feasibility of eligibility criteria
Select sites with eligible patients
Pharmacovigilance and Patient Safety
Observe safety signals (e.g., previously unknown adverse events)
Identify subpopulations with unique risk profiles
Describe safety in real-world conditions, validate or disprove safety signals
General Medical Research
Generate new hypotheses about diseases, causes, and potential new therapies
Understand real patient demographics, comorbidities, comedication
Describe the natural history of diseases
Medical Services and Health Outcomes
Understand treatment patterns and pathways
Evaluate clinical effectiveness and patient outcomes over time
Discover auxiliary benefits beyond original indication
RWD and associated RWE may constitute valid scientific evidence, depending on the characteristics of the data and the analytical methods used. It will, however, take time for RWD guidelines to be established and the quality of RWD to achieve a high enough standard so that RWE studies can be used as a hypothesis confirming method, especially in the drug regulatory process. Nevertheless, RWD can now answer very important questions for which RCTs would be too time-consuming and costly for individual sponsors to conduct.
From RWD to RWE: An Example
After more than two decades of clinical trials conducted in hypertension, it is still unclear which antihypertensive class is better as first-line therapy for stroke prevention.{14,15} As most beta blockers (BBs) and angiotensin conversion enzyme inhibitors (ACEIs) are off patent and their clinical outcome results are unpredictable, it is unlikely that a sponsor would ever conduct a lengthy and costly clinical trial to compare BBs to ACEIs in stroke prevention. However, as seen in the following, RWD can provide valuable clinical insight to this comparative research.
The population to be evaluated is defined as having the ICD-10 code for hypertensive diseases (I10 to I15) and never having any cardiovascular medication before (taking either a BB or an ACEI as a first-line therapy). The ACEI group must have an ACEI but no BBs, and the BB group must have a BB but no ACEIs. To focus on relatively recent data, only patients with start of therapy after January 2013 are included (see Figure 5).
Figure 5: Cohort Definition Comparing ACE Inhibitors to Beta Blockers for First-Line Treatment of Hypertension
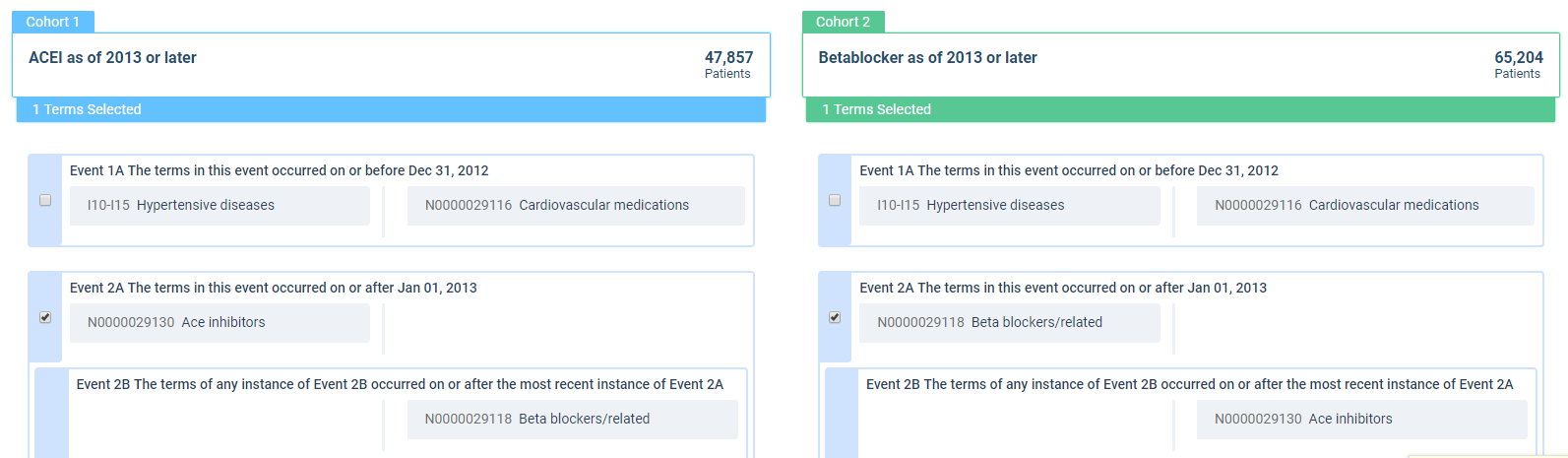
The start of therapy has been defined as the index event with a three-year observation period, starting 30 days after the index event (to exclude carryover effects from any diagnosis documented at the index date). The browser-based analytics platform then provides the results to a defined outcome (i.e., the risk of experiencing a cerebrovascular event [I60- I69] within the observation period) (see Figure 6).
Figure 6: RWD Results Comparing Risk of Experiencing Any Cerebrovascular Event Up to Three Years After Starting Antihypertensive Therapy with an ACEI or BB

Since retrospective RWD observations are not based on randomized patients, confounding factors with the potential for introducing bias must be considered. In this case, the BB group had a slightly higher percentage of patients with cardiac arrhythmias, heart failure, atrial fibrillation, and ischemic heart disease, but was otherwise comparable to the ACEI group.
There are several methods to balance for such confounding factors, with two of the most common being stratification and propensity scoring. Stratification was applied in this example, with two more analyses being performed. One subgroup contained only patients with at least one of these cardiac comorbidities; the second excluded all potential confounding cardiac comorbidities. Both strata delivered results comparable to those seen to the initial cohort, indicating that the cerebrovascular advantage of the ACEI may not be confounded by a slight imbalance in cardiac comorbidities.
More sophisticated cohort definitions and biostatistical methodologies may bring this simple example closer to a hypothesis-confirming study, rather than a hypothesis-generating one. This example also shows that the use of RWD in combination with user-friendly analytical tools can very quickly provide a picture about the therapeutic effectiveness of treatments in real medical practice, rather than in the artificial environment of an experimental and costly RCT.
Scientific Use of “Big Data” in Medical Research: General Considerations
Discussions about the use of healthcare data for research are often motivated by two fundamental questions:
- Which is the optimal source of data?
- RCTs are a very exact methodology to evaluate a treatment in isolation, under ideal conditions, and in a highly selective population. The results depend on patient eligibility criteria and on the details of the study protocol, providing insights to scientists on the effects of a molecule in development.
- RWE studies represent the collective experience from thousands of physicians treating millions of patients. They provide a complete view at the actual use of a product, its effectiveness, and related adverse events in a real medical setting. The results are influenced by treatment standards, physicians’ prescribing behavior, pharmaceutical company marketing, and documentation quality.
- A third data source is gaining more and more attention: patient-reported outcomes (PROs), including those communicated in social media. Here, the “voice of the patient” comes into play, and the information can be considered as if coming from an expanded focus group whose members are providing unfiltered, spontaneous feedback toward an overall view of the treatment’s reputation.
- While very sophisticated and scientific mentalities may still consider RCTs as the only gold standard in clinical research, one must admit that only together with RWE and PRO can a holistic picture about a treatment—its efficacy, tolerability, and effectiveness—be achieved. These three methods should not be viewed as competitive, but rather as complimentary.
- Who can use a patient’s medical data?
This question can be approached from an ethical standpoint or from a legal definition of “ownership.” The legal aspects, such as those related to privacy, data forwarding, or the analyzing process, must obviously be considered when using patient data for any research purposes. Compliance with every applicable law is a must, and the use of data created by one organization and used by another requires a contractual agreement.
The ethical and scientific views add nuance to this issue. A look at the life cycle of a study from data collection to publication shows that every step in the process adds value in advancing clinical research (see Figure 7):
- The patient allows the healthcare provider to document her or his health data
- The provider joins a data network, so healthcare information can be shared and analyzed by researchers
- These researchers analyze the data and publish the results
Only then can progress in medicine be achieved toward helping patients with the same condition.
Figure 7: Data Value Chain
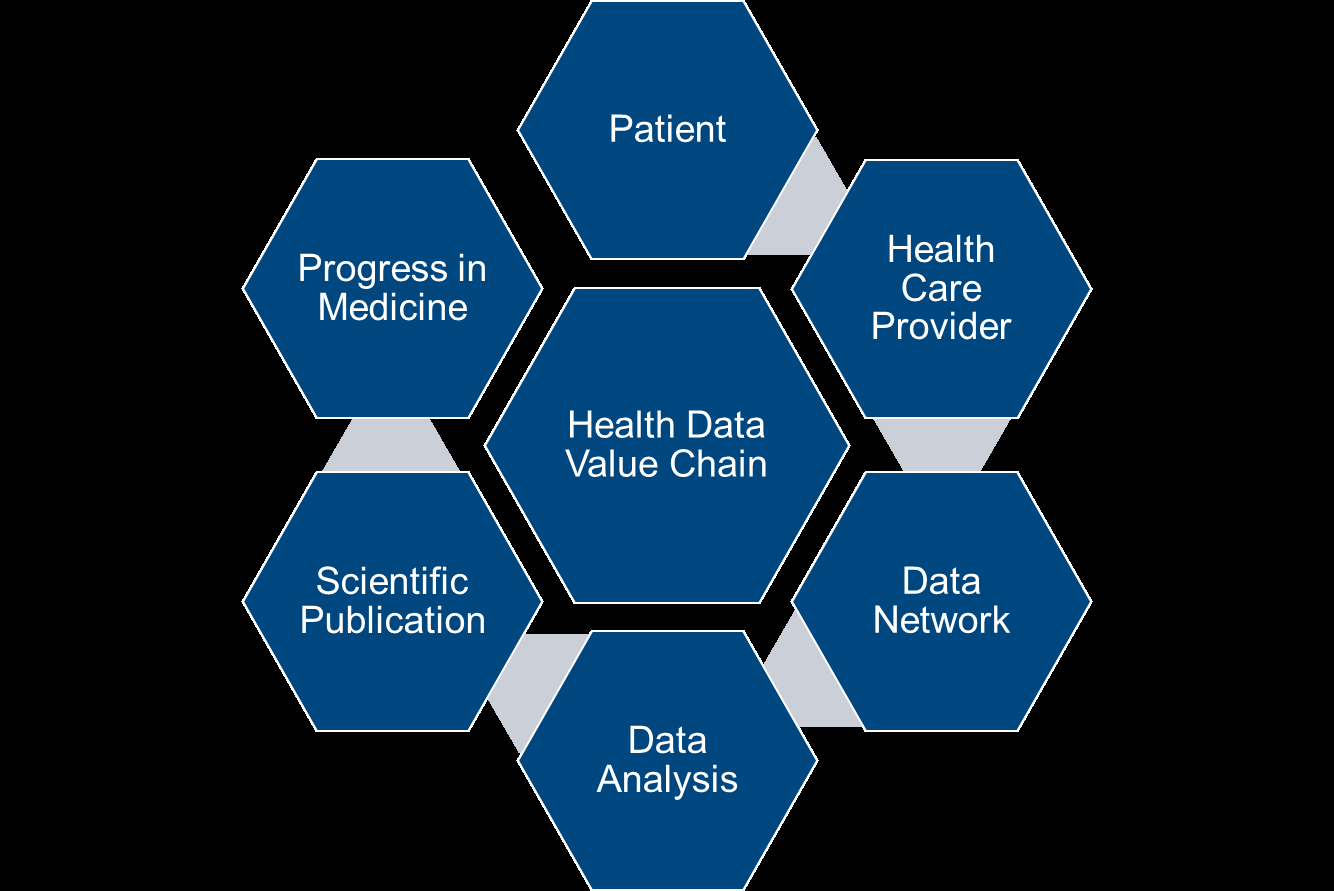
All participants and steps in this circle make valuable contributions to data-driven medical progress. Patients who will suffer due to a medical condition in the future should be able to benefit from the information obtained from those who suffer the same medical condition today. To define an ethical “data ownership” is rather difficult, but the use of patient data should be available to researchers aiming to improve how clinical research is conducted.
References
- Lamberti MJ, Chakravarthy R, Getz KA. 2016. Assessing practices & inefficiencies with site selection, study start-up, and site activation. Appl Clin Trials August 5. www.appliedclinicaltrialsonline.com/assessing-practices-inefficiencies-site-selection-study-start-and-site-activation
- Lamberti MJ, Brothers C, Manak D, Getz K. 2012. Benchmarking the study initiation process. Therap Innov Reg Sci 47(1):101–9. https://journals.sagepub.com/doi/abs/10.1177/2168479012469947
- Tufts Center for the Study of Drug Development. 2018. Rising protocol complexity is hindering study performance, cost, and efficiency. Impact Report 20(4). https://csdd.tufts.edu/impact-reports/
- The Office of the National Coordinator for Health Information Technology. Health IT Data Summaries. https://dashboard.healthit.gov/apps/health-information-technology-data-summaries.php?state=National&cat9=all+data&cat1=ehr+adoption#summary-data
- Thomas DW, Burns J, Audette J, et al. 2016. BIO Industry Analysis Reports. Clinical Development Success Rates 2006–2015. https://www.bio.org/sites/default/files/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf
- Getz K. 2014. Improving protocol design feasibility to drive drug development economics and performance. Int J Env Res Pub Health 11(5):5069–80. https://www.researchgate.net/publication/262303511_Improving_Protocol_Design_Feasibility_to_Drive_Drug_Development_Economics_and_Performance
- Getz K, Stergiopoulos S, Short M, et al. 2016. The impact of protocol amendments on clinical trial performance and cost. Therap Innov Reg Sci 1-6. https://www.mdsol.com/sites/default/files/Abstract_The%20Impact%20of%20Protocol%20Amendments%20on%20Clinical%20Trial%20Performance%20and%20Cost_2016.pdf
- Shenoy P, Harugeri A. 2015. Elderly patients’ participation in clinical trials. Perspect Clin Res 6(4):184–9. https://www.ncbi.nlm.nih.gov/pubmed/26623388
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). 2016. ICH Harmonised Guideline. Integrated Addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2). Investigator – Adequate Resources (4.2.1). https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4_2016_1109.pdf
- Bothwell LE, Greene JA, Podolsky SH, et al. 2016. Assessing the gold standard—lessons from the history of RCTs. NEJM 374:2175–81. https://www.nejm.org/doi/10.1056/NEJMms1604593
- Howie LJ, Peppercorn JM. 2014. The ethics of clinical trials for cancer therapy. NC Med J 75(4):270–3. www.ncmedicaljournal.com/content/75/4/270.abstract
- Murthy VH, Krumholz HM, Gross CP. 2004. Participation in cancer clinical trials: race-, sex-, and age-based disparities. JAMA 291(22):2720–6. https://jamanetwork.com/journals/jama/fullarticle/198896
- Berger ML, Sox H, Wilke RJ, et al. 2017. Good practices for real‐world data studies of treatment and/or comparative effectiveness: recommendations from the joint ISPOR‐ISPE Special Task Force on real‐world evidence in health care decision making. Pharmacoepidemiol Drug Saf 26(9):1033–9. https://www.ncbi.nlm.nih.gov/pubmed/28913966
- Ravenni R, Jabre JF, Casiglia E, Mazza A. 2011. Primary stroke prevention and hypertension treatment: which is the first-line strategy? Neurol Int 3(2):e12. https://www.pagepress.org/journals/index.php/ni/article/view/ni.2011.e12/0
- Hong K-S. 2017. Blood pressure management for stroke prevention and in acute stroke. J Stroke 19(2):152–65. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5466289/

Manfred Stapff, MD, PhD, (Manfred.Stapff@trinetx.com) is Senior Vice President and Chief Medical Officer at TriNetX, Inc. in Cambridge, Mass.

Jennifer Stacey (Jennifer.Stacey@trinetx.com) is Vice President of Clinical Sciences at TriNetX, Inc.


