
Clinical Researcher—January 2019 (Volume 33, Issue 1)
PEER REVIEWED
Emily Palmisano Holliday, MACPR; Mary Raber Johnson, PhD, RAC
In the past several years, clinical trials have continued to increase in number and complexity. Site infrastructure must acclimate to technology and create adaptive coping methods to meet these demands.{1} Resource adaptation within changing protocol requirements is a critical skill, particularly as technology advances in the global clinical market.{2} The streamlining of processes requires embracing new technology and new applications of current technology for best practices in training, reporting, and documentation.
The challenges and advances of the 21st century are a give-and-take system, accommodating both the increasing detail of research and providing technological solutions for excellence in safety and quality.{3} By using a risk-based approach to site operations that is rooted in technology, site staff can be supported by best practice clinical trial conduct.{4}
While current literature is abundant on risk-based monitoring application for sponsor/contract research organization (CRO)–level processes, the use of these strategies at a site level has not been fully explored. This discussion outlines essential considerations for small and/or inexperienced sites using a two-pronged approach: using technology to streamline site activities, while also reallocating resources to the areas of highest risk.
The Site Role
Implementation of a risk-based approach to site processes is key in smaller practice trials, where funding for complex software aids and specialized staff may be lacking. Within practices of one to three investigators, research support staff often maintain numerous roles to accommodate the protocol requirements. Crucial skills of a research coordinator include “…organizational skills, attention to detail, interpersonal skills, and computer and database experience…. They can make or break the clinical trial.”{5}
When communicating complex situations, site-provided details create the data story. An accurate story allows downstream data managers/statisticians to correctly categorize and interpret adverse events, relationships, and investigational product comparisons. The information must be reliable in detail and well-representative of the real-life situation.{6}
The site focus should be on crucial success factors for growing and maintaining a clinical research team: proper knowledge, practical skills, and appropriate attitude. These skills allow site staff to effectively work in teams, stay motivated for self-directing tasks, and accurately handle information.{7}
Resource Allocation in Trainings—Strategic Use of Recorded Sessions
Rather than conveying a complete spread of equal resources to each aspect of a study, a risk-based approach has been shown to improve data quality at a lower cost.{8} As outlined by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), a risk-based strategy identifies the areas of greatest risk for each specific protocol and allocates the highest priority and resources to those high-risk areas.{1,9}
It may be necessary to first discuss the purpose and rational for risk-based strategies with site staff in order to prevent negative assumptions.{10} While larger and experienced sites may afford risk-based software, smaller and/or inexperienced sites will need to tackle trainings from a simplified, risk-based perspective.
With the amount of protocol/procedural education required in clinical studies, sites should consider recording narration of slides for minor trainings and for staff self-review purposes. While major trainings may require scheduling live presenters, recording such sessions allows for simple refreshers as needed.
Creating Visibility with Documentation
It is the sponsor’s responsibility to ensure the quality of study data reported from each site. Inaccuracies or inadequacies in case report forms (CRFs) have been one of the most common reasons for FDA Warning Letters to clinical investigators.{6} Good documentation practices (GDPs) aim to prevent these types of violations by reducing/eliminating queries within the patient record. Once a site’s risk areas are identified, transparent procedures can be built into daily activities.
In novice research sites, insufficient documentation could be due to a lack of training and/or experience.{6} Examples of essential GDP training include attention paid to documentation of Good Clinical Practices, study protocols and related procedures, and standard-of-care practices onsite. Evidence of these trainings should be tracked in dedicated electronic logs or forms, to create transparency and enhance reporting.
Note the difference between the task quality and procedure of documentation. In this case, “quality” refers to training completeness, while the documentation “procedure” refers to the correctness or detail of the associated log.
Figure 1: Example of a Log with GDP Elements
Figure 1 illustrates a well-constructed log using GDP elements. Two common research adages are worthy of memorization by site staff: “What is not documented is not done” and “Document what is done, as well as what is not done.” These phrases are excellent reminders to improve the visibility, providing a preventative and ongoing feedback loop for quality management.{6}
Figure 2: Interactions Between Site Process to Ensure Quality Assurance
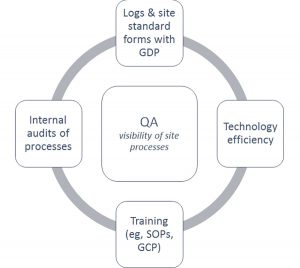
When determining the interaction between site processes, Figure 2 may provide clarity. Log/form standardization, training, and technology efficiency are dynamically involved with each other to ensure quality assurance (QA) at the site. With documentation of the research activities, specific movement of staff, patients, and data throughout the practice becomes measurable.
Consider an alternate application of technology to facilitate mandatory staff training, thereby allowing visibility within site process; for example, staff are required to review slides on a minor protocol amendment. Upon completion, staff are instructed to complete a brief electronic quiz on SurveyMonkey® to demonstrate understanding. After the training due date, the quiz results are downloaded and filed, along with the slide deck.
A QA reviewer of the quiz will report any concerns about individual results to management, and a discussion on future risk mitigation could commence—fulfilling certain safety requirements and guidelines established in 21 CFR Part 50 (Protection of Human Subjects) in the Code of Federal Regulations and in the International Council for Harmonization’s E6(R2) guideline on GCP.{11,12}
Specific strategies on documentation should be integrated into operational site processes, enabling routines to support FDA compliance. This coupling of corrective/preventative action is the best practice for efficient and high-quality site management.{1,12}
Integrating GDPs into Your Site
The specific mechanism of action to establish visibility is outlined through GDPs. Per GCPs, GDP standards should be integrated into mandatory site standard operating procedures (SOPs). The FDA ensures data quality with the acronym ALCOA, which stands for attributable, legible, contemporaneous, original, and accurate.{13} The EMA later enhanced ALCOA with four new elements (complete, consistent, enduring, and available) when considering electronic documentation, changing the acronym to ALCOA-CCEA.{9} These aspects are wholly grounded in the traceability of data origination, which are more readily examined from electronic sources.
A second document that is often required for clarity at a clinical research site is a memo- or note-to-file (MTF or NTF). These are documents included to explain errors or issues that have occurred. M/NTFs should not be used as a cover-all on deviations, but as a tool to explain exceptions to total protocol compliance. They allow for clarity on the accidental deviations that can be expected within real-world clinical trials. Additionally, original source pages or visit notes should be amended where possible to prevent unnecessary documentation.{6,14,15}
Technological Advantages within GDPs
Technology is a broad category of advancements that can be applied within various categories of clinical trials at the site level. Consider the use of paper source documents versus a low-cost, Internet-capable tablet. For the former, Figure 3 shows an example workflow for moving patient adverse event (AE)-related information reported at visits to electronic CRFs (eCRFs) in the electronic data capture (EDC) platform. The three sequential arrows represent the transcription of information from one form into another, with each having the potential for increased error rates.
Unnecessary handling of data introduces risk and increases with the number of added variables, such as the number of staff members involved or dependent actions. In this example, a clinic nurse completes the patient interview, a research assistant transcribes notes from the clinic chart to participant binder, and a study coordinator enters information into the eCRF. Within part of the workflow, an error could occur and be repeated/exacerbated in dependent steps.{16}
Figure 3: Example Workflow of Newly Reported AE, Without Application of Technology
Figure 4: Example Workflow of Newly Reported AE, With Use of Technology
Holliday_Johnson_Figures 3 and 4
Now consider Figure 4, which uses a tablet for the same scenario. The nurse is able to effectively enter the information directly while interviewing the patient. This has the minimum amount of risk, with a single transformation of data from the verbal patient report into the relevant eCRF as original data, without the need for a paper copy. Instead of a physical paper trail, the site process would be supported by the site SOP explaining this streamlined data collection process and rationale in risk mitigation.
With the tablet in use, workflow is immediately shortened by staff EDC data entry. Duplicate collection of information via secondary paper documents is removed from the equation, providing two benefits: 1) time-intensive creation/validation of paper source documents becomes unnecessary, and 2) information entry is completed only once between source and eCRF for each piece of data.
Tablet usage also allows the staff members completing patient interviews to be the ones entering data, eliminating the concern for data misrepresentation. Finally, there is also easy transportation of the exact, protocol-required eCRF entry fields, ensuring that all necessary information is collected without extraneous or unessential details.
Considerations for Technology Use in Documentation
While embracing technology, it is important to note the requirements for security when utilizing software and digital site files. Title 21 CFR Part 11 outlines the specific requirements of electronic systems and how to translate the previous requirements for physical documents into the e-version of these pages. This FDA regulation also verifies that the use of electronic records is acceptable in place of physical records. Just as hard copies of patient information must be in restricted access areas, the e-versions of trial files must be located in protected workflows, with unique, confidential passwords for each employee.
The same requirements also apply in terms of audits, such as e-files being easily accessible to the FDA. For electronic items, there should be an audit trail to provide evidence of all changes, username of the change initiator, the date, and the changes made. Unique identifying passwords can act as the signature electronically and create an appropriate imprint to verify the authenticity of the user inciting any changes or approvals. Further, records retention is essential; much of the good practices for clinical trials pertains to the accuracy and assurance of the records.{17}
Sites should also consider how systems already used onsite could be expanded for documentation in clinical trials. For example, Microsoft® Office 365® aims to comply with 21 CFR 11 requirements, and has options for audit trails and cloud-based storage.{18} AdobeTM AcrobatTM can be used for more than document viewing, and includes e-signature capabilities.{19} Identifying existing programs that show FDA compliance with electronic documentation can be a cost-saving technique to improve efficiency of technology according to GDPs.
Conclusions
Technology and risk-based considerations in site management pair productively with data management to enhance site output quality. Understanding the interaction between site activities in order to create visibility and measurability promotes risk management. Tactical application of the aspects described herein allow for optimization of site and study success in the new era of technology fluency.
References
- U.S. Department of Health and Human Services. 2013. Oversight of Clinical Investigations — A Risk-Based Approach to Monitoring, Guidance for Industry.
- Jones CT, Jester PM, Fitz-Gerald M. 2011. Issues in research management: protocol challenges in the era of complexity. Res Practitioner 12(4):121–31.
- Friedman TL. 2016. Thank you for being late: An optimist’s guide to thriving in the age of accelerations. New York: Farrar, Straus and Giroux; Chapter 3.
- Arismendy C. 2010. How to start doing office-based clinical trials. Medscape Business of Medicine. https://www.medscape.com/viewarticle/720916
- Gosnell JE. 2014. Building your clinical trial research team. In T. M. Pawlik & J. A. Sosa (Eds.), Success in Academic Surgery: Clinical Trials. London: Springer London.
- Bargaje C. 2011. Good documentation practice in clinical research. Perspect Clin Res 2(2), 59–63.
- Rajadhyaksha V. 2010. Training for clinical research professionals: focusing on effectiveness and utility. Perspect Clin Res 1(4), 117–9.
- Agrafiotis DK, Lobanov VS, Farnum MA, Yang E, Ciervo J, Walega M, Baumgart A, Mackey AJ. 2018. Risk-based monitoring of clinical trials: an integrative approach. Clin Ther 40(7):1204–12.
- European Medicines Agency. 2013. Reflection paper on risk based quality management in clinical trials. ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/11/WC500155491.pdf
- Jeong IS, Jeong JH, Hwang, YS, Youn JH. 2018. Clinical research coordinators’ attitude toward risk-based monitoring. Ther Innov Regul Sci. Epub ahead of print. doi: 10.1177/2168479018793132
- Chow S, Liu J, Ohio Library and Information Network, & Wiley Online Library. 2013. Design and analysis of clinical trials. Hoboken, N.J.: John Wiley & Sons.
- International Council for Harmonization (ICH). 2016. Good Clinical Practice: Integrated Addendum to ICH E6(R1): Guidelines for Good Clinical Practice E6(R2) https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4_2016_1109.pdf
- U.S. Department of Health and Human Services. 2016. Data Integrity and Compliance with CGMP, Guidance for Industry.
- PharmOut. 2016. How to implement good documentation practices [white paper]. https://www.pharmout.net/downloads/white-paper-how-to-implement-good-documentation-practices.pdf
- Hazra A. 2011. Use, abuse and misuse of notes to file. Perspect Clin Res 2(1), 38–40.
- U.S. Department of Health and Human Services. 2013. Electronic Source Data in Clinical Investigations, Guidance for Industry.
- U.S. Department of Health and Human Services. 2003. Part 11, Electronic Records; Electronic and Application, Guidance for Industry. https://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm125125.pdf
- Compliance Certifications for Office 365. 2018. https://products.office.com/en-us/business/office-365-trust-center-compliance-certifications
- Adobe Document Cloud. 2018. https://acrobat.adobe.com/us/en/acrobat/how-to/electronic-signatures-online-e-signatures.html

Emily Palmisano Holliday, MACPR, (emrose.palmisano@gmail.com) is a Clinical Trial Associate at Ora, Inc. in Andover, Mass.

Mary Raber Johnson, PhD, RAC, (johnson.6844@osu.edu) is an Assistant Clinical Professor at The Ohio State University.


