
Clinical Researcher—September 2019 (Volume 33, Issue 8)
PEER REVIEWED
Daniel Eisenman, PhD, RBP, SM(NRCM), CBSP
Once thought of as being pursued only in the realm of science fiction or under highly experimental conditions, the science of gene therapy is now considered to be well understood, and the field is thriving.
Gene therapy research involves delivering engineered genetic material to humans with the goal of compensating for genetic mutations, conferring the capability to produce potentially therapeutic substances, or eliciting immune responses to fight disease.
As of the writing of this article, to date, more than 2,900 gene therapy studies have been initiated worldwide.{1} Searching ClinicalTrials.gov for the keywords “gene therapy” results in more than 3,800 studies, with more than 1,000 of them currently recruiting or enrolling research subjects.{2}
The gene therapy field overcame several hurdles to reach its current state. High-profile serious adverse events have been reported over the years, including the tragic death of Jesse Gelsinger in 1999 and leukemia in children with life-threatening immune deficiencies (i.e., severe combined immune deficiency [SCID] and chronic granulomatous disease [CGD]).{3–6} These incidents occurred at the turn of the century and led to a decade of intensive study, redesign and safety testing of gene therapy technology.{7–10}
During this period, the U.S. Food and Drug Administration (FDA) issued various guidance documents regarding the manufacture of gene therapy products, design of clinical trials, and long-term follow up for certain types of investigational products for gene therapy.{11} As the field focused on reassessing safety, the number of investigational new drug applications (INDs) received by FDA for gene therapy products has grown exponentially from 1995 to 2010 (see Figure 1).{12}
Figure 1: Gene Therapy IND Applications Submitted Per Year
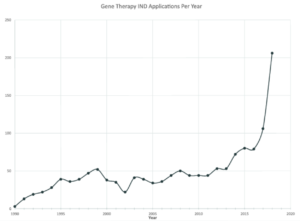
Data adapted with permission from Peter Marks, Director, FDA Center for Biologics Evaluation and Research (CBER).{12}
Since 2011, the field has regained confidence in the safety of gene therapy technology, and the number of gene therapy product INDs received by FDA has been steadily climbing; 2018 marked an all-time high number of IND applications (206), almost doubling the previous all-time high achieved in 2017 (106).
The surge in research has also led to approvals (see Table 1). FDA issued its first approval of a product containing engineered genetic material (i.e., recombinant DNA) in 2015 and has since issued six more approvals, with the most recent dated May 24, 2019. With 291 gene therapy studies currently in Phase III, several more gene therapy products will likely be considered for approval in the coming years.
Table 1: FDA Approvals of Gene Therapy Products
| Product Name | Manufacturer | Indication | Engineered Genetic Material | FDA Approval Date |
| IMLYGIC | Amgen | Melanoma | Herpes simplex virus 1 based oncolytic therapy | October 2015 |
| VAXCHORA | PaxVax | Cholera vaccine (serogroup O1) | Live, attenuated, orally administered V. cholerae bacteria, cholera toxin A gene (ctxA) deleted | June 2016 |
| KYMRIAH | Novartis | B Cell Acute Lymphoblastic Leukemia | Chimeric Antigen Receptor (CAR) T Cells, engineered with a retrovirus vector | August 2017 |
| YESCARTA | Kite (Gilead) | Non Hodgkins Lymphoma | Chimeric Antigen Receptor (CAR) T Cells, engineered with a retrovirus vector | October 2017 |
| LUXTURNA | Spark Therapeutics | Retinitis Pigmentosa | Adeno associated virus (AAV) vector delivering the RPE65 gene | December 2017 |
| DENGVAXIA | Sanofi Pasteur | Dengue serotypes 1-4 | Tetravalent dengue vaccine based on the Yellow fever 17D204 vaccine strain | May 2019 |
| ZOLGENSMA | Novartis | Spinal muscular atrophy | Adeno associated virus (AAV) vector delivering the SMN1 gene | May 2019 |
With the increasing number of late-phase gene therapy studies and continued growth of the field, it’s important for clinical research professionals to familiarize themselves with the prospects, risks, and regulatory requirements for conducting gene therapy research.
Areas of Gene Therapy Research
Approximately two-thirds of gene therapy studies are in the field of oncology.{1} With the aging Baby Boomer demographic, demand for advancements in oncology and availability of research subjects for clinical trials are likely to increase.
Common areas of oncology gene therapy research include cancer vaccines, engineered immune cells targeting cancer, and oncolytics (the use of viruses that selectively reproduce in and kill cancer cells while sparing normal, healthy cells). High-profile immunotherapies, such as chimeric antigen receptor (CAR) T cells, utilize genetic engineering to reprogram white blood cells (i.e., T cells) to specifically target cancer cells. CAR T cells have been especially successful in treating B cell malignancies in cases of resistant or refractory disease.
The second most common area of gene therapy research (11.5% of reported studies) applies to monogenic diseases.{1} While diseases caused by single gene mutations are rare, they represent low-hanging fruit from a technical perspective, as current technology offers a wealth of techniques for correcting single gene mutations. Examples of such diseases are listed in Table 2.
Table 2: Examples of Monogenic Diseases
| Disease | Description |
| Retinitis pigmentosa | An inherited form of night blindness in children that progresses to complete blindness by adolescence. |
| Severe Combined Immune Deficiency (SCID)
|
Famously characterized by the movie The Boy in The Bubble, is a disease preventing bone marrow stem cells from developing into white blood cells leaving the host immune compromised. |
| Chronic Granulomatous Disease (CGD) | Causes impaired antimicrobial activity in phagocytic cells leading to immune deficiency and granuloma formation at sites of infection. |
| Cystic fibrosis (CF) | A progressive disease leading to persistent lung infections, limiting ability to breathe as well as causing digestive problems. |
| Hemophilia A and Hemophilia B | A bleeding disorder caused by a lack of blood clotting factor VIII and IX, respectively. |
| Severe sickle cell disease | Red blood cells contort into a sickle shape and die prematurely causing anemia and blockage of vasculature. |
The third most common type of research (6.3% of reported studies) occurs in the field of infectious diseases.{1} Genetic engineering allows new avenues for development and manufacture of vaccines. FDA has already approved vaccines containing engineered genetic material to protect against cholera and dengue virus infections. Genetically engineered Ebola vaccines are undergoing evaluation in the current outbreak in the Democratic Republic of the Congo, with promising early results.{13–14}
Managing Risks Associated with Genetic Engineering and Gene Therapy Research
Most clinical researchers are familiar with the regulatory requirements pertaining to the FDA phases of research, as well as with institutional review board (IRB) review. Gene therapy studies may require additional review to assess the risks associated with the engineered genetic material, especially as the technology frequently utilizes genetically engineered viruses to deliver genetic information into target cells. Viral infection involves the transfer of the virus’ genetic material to host cells, making viruses ideal tools for gene transfer—once the viral genes responsible for viral replication and disease are removed. While genetically modified viruses have a greater safety profile than the naturally occurring unmodified variety, they remain infectious and capable of posing risks.
National Institutes of Health (NIH) Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines) provide the standard for oversight of research involving genetic engineering and gene therapy.{15} NIH Guidelines are promulgated by the NIH Office of Science Policy (OSP) and call for local oversight at the research site by institutional biosafety committees (IBCs) that report to the NIH OSP. IBCs are charged with protecting study personnel, the community, and the environment from exposure to engineered genetic material. An IBC may also advise the IRB to aid in assessing risks to the study subjects.
The requirement for IBC review applies to gene therapy research at sites that are receiving funding from the NIH or that have ever participated in NIH-funded research. Sponsors or sites that have received any NIH funding are obligated to comply with IBC review regardless of whether the funding is associated with the gene therapy study. Studies and sites completely independent of NIH funding may still require IBC review if the research and development that led to the investigational product was funded by NIH. Even if there are truly zero NIH funds involved, IBC review is considered a best practice: NIH Guidelines state that “individuals, corporations, and institutions not otherwise covered by the NIH Guidelines are encouraged to adhere to the standards and procedures set forth” in the Guidelines (Section IV-D-1).
IBCs are composed of at least five members, including at least two unaffiliated community members, who collectively possess the expertise to assess the risks associated with proposed research projects. IBC review involves assessing the risks associated with the genetically modified investigational product, as well as the adequacy of a facility’s safety practices and training intended for use of the investigational product at the site.
The IBC ensures the site has adequate incident reporting and response plans in place to address potential occupational exposures, spills, or environmental releases of the investigational product. The IBC may review informed consents and other research subject training materials to mitigate possible risks to casual or close contacts in the community. Reviewing the site’s plans for disposal of the investigational product and associated biomedical waste allows the IBC to ensure environmental protection.
Because they are charged with protecting the local community and environment, IBCs are locally based at the research site and can only oversee research at that location. For the sake of efficiency, however, local IBC meetings can be centrally coordinated and synchronized, with each IBC remaining responsible for local review.
Pharmaceutical companies and contract research organizations conducting multisite clinical trials may benefit from efficiencies provided by centrally administered IBC reviews, utilizing a model similar to central IRB reviews. NIH refers to such review bodies as externally administered IBCs and allows sites to utilize them as long as the IBCs are registered with the NIH OSP as representing the individual sites at the time the review is performed. NIH allows sites to register multiple IBCs, so an academic institution with an existing IBC focused on preclinical research in the laboratory setting may choose to utilize an externally administered IBC for review of clinical trials and industry-sponsored research.
At a single meeting, the centrally administered IBCs can conduct reviews for multiple sites participating in a single study, as long as the required local representatives are present at the meeting and separate minutes are recorded for each site. This approach streamlines the process for submission and review, providing a single point of contact for submissions as well as harmonized forms, policies, and procedures across sites.
IBCs run by institutions may have set monthly or quarterly meeting schedules, and can insert IBC review as a “blocking review” (i.e., the institution’s IRB will not accept a protocol for review until the IBC has issued its approval). Centralized IBCs, in contrast, often convene “on demand,” which eliminates the practices of submission deadlines and standing meeting schedules, and results in turnaround times measured in days instead of weeks or months. For example, in 2019 the central IBC service provided by Advarra, a commercial research compliance organization, has averaged turnaround times of 7.1 business days (N = 40 studies) from submission to review for National Cancer Institute–designated cancer centers. These centers previously experienced turnaround times of up to three to four months when working with their locally administered IBCs.
Evolving Federal Oversight for Genetic Engineering and Gene Therapy Research
NIH Guidelines have relied on advisory committees to review emerging research and advise the NIH Director regarding policy matters.{16} The role of this advisory committee has evolved over time.
In the 1980s, the ability to transfer engineered genetic material into humans was completely new to the clinical setting. Review of gene therapy studies was included in the responsibilities of the NIH Recombinant DNA Advisory Committee (RAC), which created the first set of requirements for review of gene therapy research in 1985.
Over time, FDA became increasingly involved with review of gene therapy studies and, in 1991, issued its own guidance document regarding review of gene therapy studies. By 1995, FDA and NIH agreed FDA would assume primary responsibility for review of gene therapy studies, with NIH and FDA jointly determining which studies required RAC review. The NIH Guidelines were revised in 2016 to require the IRB and IBC at the initial site conducting a gene therapy study to determine whether to recommend RAC review. This recommendation was based on the novelty of the science and whether the risks or possible toxicities were difficult to ascertain.
The latest version of the NIH Guidelines (issued on April 25, 2019) removes the requirements for IRB and IBC RAC determination, as well as protocol registration and reporting requirements to the NIH OSP. These requirements were deemed to be duplicative with the level of oversight currently provided by FDA as well as the IRB and IBC. As the RAC no longer reviews individual clinical trials, the role of the RAC was revised to focus on the “scientific, safety, and ethical issues associated with new and emerging biotechnologies.”{17}
To further emphasize this more general role, the RAC has been renamed the Novel and Exceptional Technology and Research Advisory Committee (NExTRAC). The committee’s amended charter charges it with advising the NIH Director “on matters related to the conduct and oversight of research involving emerging technologies in biomedical science….”{18}
As the committee no longer reviews large volumes of gene therapy studies, the NExTRAC can now focus on emerging biotechnology requiring the focus of a national panel of experts. Many issues are likely on the horizon for the NExTRAC, such as emerging developments in gene editing technology like CRISPR and zinc finger nucleases, especially as they pertain to clinical research. Other likely matters of future discussion include applying gene editing technology to human reproduction and environmental issues, such as gene drives and environmental release of modified mosquitos incapable of transmitting diseases.
FDA and the Future Oversight of Gene Therapy
As the lead regulatory agency for review of gene therapy studies, FDA has taken a number of steps in recent years to assist in bringing gene therapy from the realm of scientific theory and research to the world of approved and licensed therapeutics for clinical use:
- FDA has issued guidance documents for the manufacture of gene therapy products, design of clinical trials, and long term follow up (see Figure 2).{11}
- The 21st Century Cures Act authorized FDA to create the Regenerative Medicine and Advanced Therapies (RMAT) designation to allow for expedited review of regenerative medicines and advanced therapies. The associated guidance issued by FDA stated the RMAT designation also applies to “…gene therapies, including genetically modified cells, that lead to a durable modification of cells or tissues.”{19}
- FDA is expanding its capabilities to review gene therapy studies in order to accommodate the growing field. In a June 2018 interview, then-FDA Commissioner Scott Gottlieb disclosed that he expects the agency to have approved 40 gene therapies by 2022, a gargantuan number considering only four approvals had been issued at the time.{20} In a statement{21} issued in January 2019, Gottlieb and Center for Biologics Evaluation and Research Director Peter Marks mentioned plans for:
-
-
- Hiring 50 additional clinical reviewers for cell and gene therapy
- 200 IND applications for gene therapy products submitted per year by 2020
- 10 to 20 gene therapy approvals per year by 2025
-
Figure 2: FDA-Issued Gene Therapy Guidance Documents
|
In their joint statement, Gottlieb and Marks write that the growth in gene therapy “…reflects a turning point in the development of these technologies and their application to human health…. It’s similar to the period marking an acceleration in the development of antibody drugs in the late 1990s, and the mainstreaming of monoclonal antibodies as the backbone of modern treatment regimens.”{21}
Concluding Remarks
Once considered highly experimental and limited to early-phase studies at highly specialized research sites, the field of gene therapy has progressed to mainstream clinical research. With the current research boom and progression into large multi-site clinical trials, clinical researchers are increasingly likely to be involved in gene therapy studies throughout their careers.
Clinical researchers should be aware of the maturation of this field and consider the opportunities it may provide to their careers and their patients. Understanding the history and fundamental technical principles of this science will allow clinical researchers to understand the capabilities and limitations of this exciting and innovative field. It is also important for clinical researchers to be familiar with the prospects, risks, and regulatory requirements to be able to safely conduct gene therapy research.
References
- Gene therapy clinical trials worldwide. J Gene Med. https://www.wiley.com/legacy/wileychi/genmed/clinical/
- National Institutes of Health. ClinicalTrials.gov. clinicaltrials.gov
- Marshall E. 1999. Gene therapy death prompts review of adenovirus vector. Science 286(5448):2244–5.
- Stolberg SG. 1999. The biotech death of Jesse Gelsinger, The New York Times Magazine.
- Marwick C. 2003. FDA halts gene therapy trials after leukemia case in France. BMJ
- Marshall E. 2002. Gene therapy with retroviruses halted. Science.
- Worth T, Parker N, Herttualla S. 2013. History of gene therapy. Gene 525:162–9.
- Seymore LW, Thrasher AJ. 2012. Gene therapy matures in the clinic. Nature Biotech 30(7).
- David R., Doherty A. 2017. Viral vectors: the road to reducing genotoxicity. Tox Sci 155(2):315–25.
- Dunbar C, et al. 2018. Gene therapy comes of age. Science 359(175).
- U.S. Food and Drug Administration. Cellular and gene therapy guidances. https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/default.htm
- Marks P. FDA gene therapy IND applications submitted per year. Data adapted with permission from Peter Marks, Director, FDA Center for Biologics Evaluation and Research (CBER).
- National Institute of Allergy and Infectious Diseases. Ebola vaccine webpage. https://www.niaid.nih.gov/diseases-conditions/ebola-vaccines
- University of Minnesota Center for Infectious Disease Research and Policy. 2019. Ebola cases climb by 44 as vaccine trial affirms high efficacy. http://www.cidrap.umn.edu/news-perspective/2019/04/ebola-cases-climb-44-vaccine-trial-affirms-high-efficacy
- National Institutes of Health. NIH guidelines. https://osp.od.nih.gov/biotechnology/nih-guidelines/
- Oversight and Review of Clinical Gene Transfer Protocols: Assessing the Role of the Recombinant DNA Advisory Committee. Historical and Policy Timelines for Recombinant DNA Technology. Copyright 2014 by the National Academy of Sciences.
- Collins FS. Statement on modernizing human gene therapy oversight. https://www.nih.gov/about-nih/who-we-are/nih-director/statements/statement-modernizing-human-gene-therapy-oversight
- Amended Charter, Novel and Exceptional Technology and Research Advisory Committee (Formerly Recombinant DNA Advisory Committee). 2019. https://osp.od.nih.gov/wp-content/uploads/NExTRAC_Charter_041219_508.pdf
- U.S. Food and Drug Administration. Expedited programs for regenerative medicine therapies for serious conditions. https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm585414.pdf
- Terry M. Gottlieb at BIO 2018: 40 gene therapy approvals by 2022. Biospace. https://www.biospace.com/article/gottlieb-at-bio-2018-40-gene-therapy-approvals-by-2022/
- U.S. Food and Drug Administration. Statement from FDA Commissioner Scott Gottlieb, M.D. and Peter Marks, M.D., Ph.D., director of the center for biologics evaluation and research on new policies to advance development of safe and effective cell and gene therapies. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm629493.htm
Daniel Eisenman, PhD, RBP, SM(NRCM), CBSP, (daniel.eisenman@advarra.com) is Director of Biosafety Services for Advarra in Research Triangle Park, N.C.
The author would like to acknowledge the assistance of Shaun Debold, Stephanie Pyle, Cameron Stahl, and Sarah Bowman in the preparation of this manuscript.


