
Clinical Researcher—January 2021 (Volume 35, Issue 1)
PEER REVIEWED
Wasi Akhtar, BPharm, MBA
Biosimilars, even though they are newer versions of existing, trade-name biological products whose patents have expired and share identical amino acid sequences with those earlier products, are not identical to the reference product. Biosimilars do not utilize the same living cell lines, production processes, or raw materials{1} as the innovator drugs (the reference originator biologics).
As novel drug development expands in the 21st century, biologics are leading the way, yet they correspond to the costliest of treatments. It is anticipated that using biosimilars will lead to an estimated $54 billion reduction in direct spending on biologic drugs from 2017 to 2026 (all monetary statistics in this article are in U.S. dollars).{2}
At present, the total number of biosimilars approved by the U.S. Food and Drug Administration (FDA) is 28, with Hulio being the most recent approval.{3} The FDA’s support of biosimilars has instilled confidence among pharmaceutical companies to pursue their development as a positive trend for both consumer needs and corporate viability.
This article provides insight into the guidelines issued by the FDA regarding considerations related to biosimilars development. Important considerations include the role of data analysis and a focus on such key concepts as the totality of evidence, data requirements, immunogenicity, and interchangeability.
SIDEBAR:
A Reference product is a single biological product, already approved by the FDA, to which a proposed biosimilar product is compared.
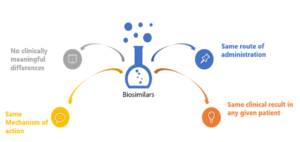
A Biosimilar is a biological product that is highly similar and has no clinically meaningful differences from an existing FDA- approved reference product.
An Interchangeable product is a biosimilar product that can be substituted for the reference product without the intervention of the prescribing healthcare provider.
Source: https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products
Key Attributes of Biosimilars as Related to Biologics
Background
Biologics (also known as genetically engineered or biotech products) are a class of medications produced from living cells using recombinant techniques. This class of medication is comprised of large molecules with a complex structure that includes a primary amino acid sequence, higher order secondary and tertiary structures, and various post-translational modifications.
A biosimilar is a “highly similar” biological product to one that has been previously approved by the FDA, and shall have no clinically meaningful differences in terms of safety, efficacy, and purity; however, there can be few minor changes in terms of active ingredients. The biosimilar product should have an identical route of administration, strength, and dosage form as the earlier product and, like all FDA-approved products, must comply with Good Manufacturing Practices demonstrating drug quality.{4}
Definitions from the FDA, European Medicines Agency, and World Health Organization{5}

While Europe revolutionized the development and medicinal applications of early biologic products such as vaccines and antitoxins, the United States has been leading the innovations in biotechnology and biologic therapies in the 21st century.
The Patient Protection and Affordable Care Act of 2010 allows the approval of biosimilars in the U.S. and allows certain clinical and nonclinical requirements for drug approval to be waived if regarded as unnecessary by the FDA.{6}
The Evolution of Biosimilars in the U.S.{7}

Methods
This clinical perspective overview was performed by analyzing the FDA’s regulatory policies, guidance documents, and related information for the biosimilar pathway, as well as by reviewing related literature and opinions from publicly available websites.
Trends and the Territory for Biosimilars
According to Grand View Research, Inc., the biosimilar market is expected to grow at a CAGR of 34.2% and attain a global value of $61.47 billion by 2025. The market for biosimilars in the U.S. is growing at a steady pace owing to high drug costs and production timelines.{8}
Unlike the case for generic drugs, for biosimilars there is an abbreviated pathway for approval that must validate that they are highly similar to the reference biologic and that there are no meaningful differences from the clinical perspective. There is a concept of interchangeability, by which the FDA means a product (with an interchangeable designation) can be replaced with the reference biologic without the intervention of the prescriber.{9} The “high similarity” between the proposed biosimilar and biologic (reference product) must be demonstrated.{10}
The production of biosimilars is a complex, multi-step procedure; at each stage, such factors as the production cell line, culture conditions, and formulation may alter the final product through post-translational modifications. Since biologics and biosimilars are created in living cells, they cannot be chemically synthesized like generic drugs.
An Abbreviated Biologics License Application (aBLA) to FDA for the proposed biosimilar should include information demonstrating biosimilarity, particularly the data derived from the analytical studies for clearly proving and demonstrating “high similarity” to the reference biologic.{11}
FDA Approval of Biosimilars
The Biologics Price Competition and Innovation Act (BPCIA) of 2010, through the abridged approval pathway for biosimilars, allows approvals in fewer steps as compared to the reference product. However, this certainly does not mean that lower standards have been adopted by the FDA for the abbreviated pathway, as the producers of biosimilars should furnish extensive data packages that meet the stringent standards determined by the agency.
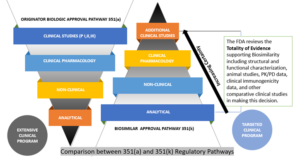
The assessment of biosimilars is performed on a case-dependent basis, and each application’s data requirements will vary accordingly. Typically, the FDA considers the following types of data while assessing a biosimilar:
Analytical Studies—To illustrate the molecular profile of the biosimilar in a manner showing high similarity to the reference product, both from structural and functional perspectives.
Animal Studies—To evaluate toxicity of the biosimilar.
Clinical Pharmacology Studies—To give proof of evidence in terms of safety, quality, and efficacy of the biosimilar (may include pharmacokinetic (PK) and pharmacodynamic (PD) assessments).
Additional Clinical Studies of Biosimilarity—The objective of a biosimilar development procedure is to validate high similarity between the biosimilar and reference product, rather than separately establishing the safety and efficacy of the proposed product.
Totality of Evidence
The FDA has a very robust approach toward the evaluation of biosimilarity called “Totality of Evidence,” which is aimed at comparative testing and approvals (depicted below). The agency advises the developers of biosimilar candidates to take a multi-step approach and, at each step, to compare the candidate to the biologic (reference product), evaluate it in terms of where there may be residual uncertainty, and perform studies aimed at mitigating those uncertainties. Each step in the biosimilar approval pathway should decrease residual uncertainties from the previous stage.{12}
Comparison Between Innovator and Biosimilar Regulatory Pathways{13}
Data Requirements for Development of Biosimilars
A biosimilar application should demonstrate biosimilarity by providing evidence that the proposed product has highly similar characteristics to the reference product, based on thorough analysis of the reference product and sequential testing of the biosimilar. The major development and application fundamentals are summarized below:
- Design controls, validation, and verification studies
- Biosimilar development through quality by design approach
- Analytical similarity through statistical data
- Clinical aspects
- FDA Guidance on Biosimilar Labeling
Demonstration of Biosimilarity from Clinical Pharmacology Data
This typically involves three key concepts—exposure and response assessment, evaluation of residual uncertainty, and assumptions about analytical quality and similarity.
While determining the safety, efficacy, and purity of any biological product, it is essential to evaluate the “exposure and response” along with a thorough assessment to ascertain any possible clinically meaningful difference between two products. The response is a precise measure of the pharmacological aspects in relation to effectiveness and adverse reactions.{14}
Immunogenicity and Safety Assay
This assay describes the generation of the immune response within the body to a biotherapeutic that may result in immune-mediated toxicity and/or a lack of effectiveness. Biologic drug treatments introduce a foreign substance, in response to which anti-drug antibodies (ADAs) may form. Due to this, there can be serious safety and efficacy implications for biosimilar drug programs; for example, ADAs may block the functionality of the biosimilar, greatly alter the PK of the biosimilar in a biologic system, or even cause acute and long-term health consequences. In such cases, it might not be suitable for additional studies to be conducted, largely depending on the extent of such potential safety and efficacy concerns.{15}
At least 36 publications have presented primary evidence explaining the effectiveness of biosimilars that followed on from major biologics with proteins 200 amino acids in length or greater (including etanercept, adalimumab, infliximab, and rituximab). ADAs were tested in 24 experiments considering larger biosimilars, and seven provided details on neutralizing antibodies (NABs).
Among the smaller biosimilars, 13 studies measured ADAs and four presented NABs (erythropoietin, filgrastim, human growth hormone). In all the studies documenting immunogenicity results, ADA and NAB levels were found to be comparable across all disease indications and treatment groups at baseline and at the end of the study, the authors add.{16}
Trial Designs for Developing Data Regarding Biosimilars
A crossover design is acceptable, if possible, for PD studies using products with a short half-life (e.g., less than five days), a rapid PD response, and a low incidence of immunogenicity. However, this type of clinical trial is most sensitive to PK assessment of similarity.
A parallel design would typically be needed for products with a longer half-life (e.g., more than five days) or for which recurring exposures may lead to an increased immune response, thereby effecting the PK/PD assessments to derive similarity. Scientific rationale for the choice of the research dose (e.g., one or several doses) and route of administration should be provided by the sponsors.
Population Type to Use for Study
PK/PD studies to demonstrate similarity can be performed with healthy volunteers, as this practice is often considered to deliver more sensitivity in the results and as being likely to produce less variability in PK values as compared to patients with underlying diseases and associated medications. However, if safety and other considerations prohibit the involvement of healthy volunteers, the clinical pharmacology studies can be conducted in patients.{17}
Dose
The appropriate dose that can provide clinically significant and understandable data should be chosen. For example, in scenarios where the studies are performed in a patient population, the standard dose for the reference biologic product might be the suitable choice, as this might best determine the pharmacological effects in a clinical setting.
Route of Administration
When conducting in-human PK and PD studies, the route of administration for the proposed biosimilar product should ideally be the same as for the reference product.{18}
PK Measurement
In the case of a single-dose study, the total exposure must be calculated as the area under the biological product concentration-time curve from time zero to time infinity; however, in the case of multiple-dose studies, the measurement of total exposure must be the area under the concentration-time profile from time zero to time tau over a dosing interval at steady-state.{19}
Extrapolation of Evidence on Effectiveness and Safety to Other Indications
The safety and efficacy of biologics should be determined in clinical trials in order to gain approval for each clinical use or indication sought. Extrapolation is the approval of a proposed biosimilar product in one or more additional indications for which the reference biologic is licensed, whereas the biosimilar has not been studied in clinical trials.
There are some items that the FDA says should be scientifically justified when considering extrapolation of signs and symptoms. The first is that the mechanism of action in the state of use—including the target/receptor for each biosimilar activity/function, binding, dose/concentration reaction, molecular signal pattern for target receptor involvement, and relationship between the biosimilar structure and target/receptor interactions and target/receptor position and expression—should be the same.
Extrapolation is based on all the evidence available in the biosimilar application, previous protection and efficacy results accepted by the FDA for other licensed reference product indications, and the understanding and evaluation of different scientific factors for each reference product.
Indication extrapolation reduces or removes the need for some indications of interest to already have been approved for the reference product when studying the potential biosimilar in clinical trials. This concept is crucial to achieving the goals of abbreviated approval pathways for biosimilars at a substantially lower cost. Some of the characteristics that may be considered for extrapolation are summarized below:
- Mechanism of action in each condition
- Binding and molecular signaling
- Location and acceptance of target/receptor
- PK and biodistribution
- PD methods may also provide important mechanism of action information
- Expected toxicities
- Differences may exist in each condition of use and patient population
Interchangeability
Interchangeability is a subset of biosimilar products defined within the statute, which basically means the biosimilar product can be substituted for the reference biologic product without the intervention of the prescriber. It is expected that the biosimilar will provide the same clinical result as the reference product in any given patient. Additionally, if it is a multi-use product (products that are administered more than once), switching or alternating between the proposed interchangeable and the reference biologic product should not increase the risk of safety or diminished efficacy compared with using the reference biologic product multiple times.{20}
Key Attributes of an Interchangeable

Conclusion
The FDA has implemented legal frameworks for authorizing the development and marketing of biosimilar medicinal products. Based on the FDA guidance, comparative clinical safety and efficacy data will be necessary if there are residual uncertainties about the biosimilarity of the two products being compared.
Biosimilar product development follows a stepwise approach for determining the similarity of a reference biologic and proposed biosimilar. Clinical pharmacological studies play a crucial role in demonstrating biosimilarity and involve microbial and chemical analyses, in vitro biological patency assay, in vivo toxicological studies, and human clinical studies.
To determine biosimilarity of the proposed product to a reference biologic, the clinical pharmacology data are extremely important. PK and PD data are critical to support assertions of the clinical similarity between the biosimilar product and the reference product. An exposure-response assessment can significantly abbreviate the clinical development pathway of a biosimilar. PK/PD studies may replace a Phase III therapeutic equivalence study for biosimilars.
Resources
Questions and Answers on Biosimilar Development and the BPCI Act Guidance for Industry. https://www.fda.gov/media/119258/download
Biosimilars: Licensure for Fewer Than All Conditions of Use for Which the Reference Product Has Been Licensed. https://www.fda.gov/media/134932/download
Biosimilars: Comparative Analytical Assessment and Other Quality-Related Considerations Guidance for Industry. https://www.fda.gov/media/125484/download
Hunter SA, Cochran JR. 2016. Cell-Binding Assays for Determining the Affinity of Protein-Protein Interactions: Technologies and Considerations. Methods Enzymol 580:21–44. doi: 10.1016/bs.mie.2016.05.002
Alten R, Cronstein BN. 2015. Clinical Trial Development for Biosimilars. Semin Arthritis Rheum 44(6 Suppl):S2–8. doi:10.1016/j.semarthrit.2015.04.002. https://pubmed.ncbi.nlm.nih.gov/26058550/
Reinisch W, Smolen J. 2015. Biosimilar Safety Factors in Clinical Practice. Semin Arthritis Rheum 44(6 Suppl):S9–15. doi:10.1016/j.semarthrit.2015.04.005. https://pubmed.ncbi.nlm.nih.gov/26058551/
Isaacs J, Gonçalves J, Strohal R, Castañeda-Hernández G, Azevedo V, Dörner T, McInnes I. The Biosimilar Approval Process: How Different is it? https://considerations.bmj.com/content/1/1/3
Heinemann L, Khatami H, McKinnon R, Home P. 2015. An Overview of Current Regulatory Requirements for Approval of Biosimilar Insulins. Diabetes Technol Ther 17(7):510–26 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4504437/
References
1. Declerck P, Farouk-Rezk M, Rudd P. 2015. Biosimilarity Versus Manufacturing Change: Two Distinct Concepts. Pharmaceutical Research 33. 10.1007/s11095-015-1790-3.
2. Mulcahy AW, Hlavka JP, Case SR. 2018. Biosimilar Cost Savings in the United States: Initial Experience and Future Potential. Rand Health Q 7(4):3. PMID: 30083415; PMCID: PMC6075809.
3. https://www.fda.gov/drugs/biosimilars/biosimilar-product-information
4. https://fas.org/sgp/crs/misc/R44620.pdf
5. https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products (FDA)
https://www.ema.europa.eu/en/human-regulatory/overview/biosimilar-medicines-overview (EMA)
https://www.who.int/bulletin/volumes/96/4/17-206284/en/ (WHO)
7. http://gabionline.net/Reports/The-evolution-of-biosimilars-in-the-US
8. https://www.grandviewresearch.com/press-release/global-biosimilars-market
9. https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products
10. https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products
11. https://www.fda.gov/media/119258/download
12. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. https://www.fda.gov/media/82647/download
13. https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval
14. https://www.fda.gov/media/82647/download
15. Krishna M, Nadler SG. 2016. Immunogenicity to Biotherapeutics—The Role of Anti-drug Immune Complexes. Front Immunol 7:21. doi:10.3389/fimmu.2016.00021
18. https://www.fda.gov/media/88622/download
20. https://www.fda.gov/media/82647/download

Wasi Akhtar, BPharm, MBA, specializes in Presales Leadership and Strategy—Regulatory Affairs for Freyr Solutions in Bengaluru, Karnataka, India.


