
Clinical Researcher—June 2024 (Volume 38, Issue 3)
TRIALS FOR TODAY
Brian Finrow, JD
The antimicrobial therapy world is undergoing a pivotal shift toward prevention.{1} Preventive antimicrobials—in the past, an underexplored concept—are a promising solution to the antibiotic resistance problem. This also represents a paradigm shift in healthcare management—better for patients, payers, and healthcare service providers alike.
This shift is driven in part by recent advances in cell engineering technology{2} and the important safety advantages of biologic drugs over their small-molecule cousins. However, not all of the barriers to wider adoption are scientific or biological—this novel therapeutic modality has important clinical, regulatory, and patient recruitment implications.
Commercial Advantages of Preventive Antimicrobials
The market dynamics of preventive antimicrobials differ significantly from conventional treatments (antibiotics, in most cases). This is a good thing for would-be drug developers, as the market incentives for developing novel antibiotics are famously broken.{3}
In any given disease, the preventive market invariably outstrips the treatment market in size. This is due to the broader base of potential users—essentially, anyone at risk of contracting the disease. The larger total addressable market (TAM, in Silicon Valley’s jargon) for preventive drugs means a broader scope for revenue generation, and a healthier investment case to attract the capital needed to fund development. A larger TAM goes a long way to solving the broken incentives faced by conventional antibiotics (see Figure 1).
Figure 1: The market for a CDI preventive is sensitive to the product’s price, safety, and ease of administration
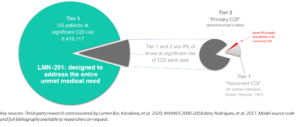
Preventive drugs also offer second rationale to support investment, in that they are generally a superior value proposition for patients. This is for obvious reasons—everybody would prefer to avoid a disease in the first place than receive life-saving care after symptom emergence. Since many infections require hugely costly healthcare services to manage, prevention can also generate savings for the healthcare system itself. These are not small numbers—dealing with just one hospital-associated disease, C. difficile infection (CDI), is estimated to comprise 2.3% of all U.S. hospital spending.{4} This amounts to billions of dollars lost to the U.S. economy annually. If you also factor the indirect costs of CDI (premature mortality and morbidity, burden on caregivers and family), the total costs are far larger.
In short, the potential financial rewards for preventive antimicrobials are superior to the daunting economics facing conventional antibiotics. This creates an encouraging opportunity to create a new generation of antimicrobials without undue reliance on public subsidies.
Regulatory and Clinical Development Considerations
These improved commercial prospects do create some unique challenges for regulatory and clinical development, however. Most importantly, the larger TAM comes from the fact that preventive drugs, by definition, are given to people at risk of the disease, which is always a larger number than the number actively suffering the disease. In turn, this implies larger clinical trials. Trial size is a major driver of development costs, so this must be confronted honestly when building a business case for a preventive antimicrobial.
There are also regulatory implications. Statistically, the preventive approach involves treating many to benefit a few, raising the bar for safety. Put into health economics terms, the larger TAM translates directly into a higher “number needed to treat” (NNT in the jargon of health economists). The NNT for any preventive drug will always be larger than for an analogous treatment for the same disease.
In turn, a higher NNT effectively raises the bar for safety and tolerability since the product will be used by many healthy people who have not (yet) contracted the disease. Regulators therefore scrutinize these drugs more stringently, necessitating comprehensive trials to demonstrate a favorable risk-benefit ratio. Such trials must be designed meticulously to balance the need for robust data against ethical considerations and practical feasibility. Relatedly, if the TAM and NNT get too large, this can generate concerns about Good Manufacturing Practices production costs, since so many doses must be delivered for each avoided harm.
Resolving the Contradictions
Addressing these challenges requires clear thinking, but fortunately, straightforward solutions do present themselves when looking at certain scenarios.
High-Risk Periods
For example, there are known high-risk periods for many infections, and it’s not hard to identify patients during these times through existing disease surveillance tools. CDI is a great example—it is by far the most common healthcare acquired infection and in most cases is caused by antibiotics. Usually prescribed for something completely unrelated (post-operative infection, for example), many antibiotics kill off the commensal bacteria in the gastrointestinal tract that ordinarily protect us from CDI. This well-documented, close relationship to certain antibiotics makes it very easy to identify the high-risk period from billing codes, since the diagnosis code predictably follows the codes for certain antibiotics prescriptions. This makes it relatively straightforward to find and enroll patients who stand to benefit. Identifying the risk period for traveler’s diarrhea is even easier: one can simply recruit travelers to regions endemic the major pathogenic enteric bacteria, especially enterotoxigenic E. coli and Campylobacter jejuni (Africa, India, and Central America).
In short, it is not necessary to recruit randomly. Most infections have known risk periods, and it is usually straightforward to recruit individuals because they (or their physicians) are often fully aware of that fact.
Trial Enrichment Opportunities
The U.S. Food and Drug Administration’s (FDA’s) trial guidance document on trial enrichment is a second good source for inspiration for preventive drug trial design. Again, C. difficile is instructive. CDI incidence varies by antibiotic, the nature of the preceding infection for which the causal antibiotic was used, and the health status of the patient.
For example, clindamycin prescribed for an unrelated infection carries a >10% risk,{5} whereas the risks can exceed 50% in the case of antibiotics prescribed for CDI itself in individuals who have experienced multiple prior CDI recurrences.{6} To a first approximation, this translates into a five-times smaller trial in the latter population while holding statistical power constant. While label negotiations are a distinct activity, the FDA’s guidance makes clear that enriching the study population in this manner will not necessarily limit the scope of the label if the underlying biological mechanism is understood.
Seres Therapeutics recently pursued exactly this strategy in developing Vowst, its fecal microbiota transplant product for CDI recurrence prevention. Seres’ pivotal trial enrolled individuals with at least three prior CDI recurrences, enriching their primary endpoint event rate by double (from a more typical CDI recurrence rate of 25% percent to something closer to 50%). Nevertheless, the FDA approved a label for the somewhat broader universe of patients suffering their first recurrence.{7}
This idea can be taken too far, however. While increasing statistical power, enrichment can also shrink the recruitable trial population so far that it becomes impossible to find enough patients. If taken too far, it can affect the external validity of the study results and therefore the breadth of the market label that the FDA is ultimately willing to approve.
Nevertheless, the general principle is sound—by focusing on individuals at heightened risk during a defined period, trials can yield more meaningful data while simultaneously decreasing costs and minimizing the number of patients exposed to the investigational drug, delivering a triple win.
Clinical Trial Efficiency
A third area for creative thought is clinical trial efficiency, particularly in terms of designing clinical studies that prioritize participant and clinician experience. This involves treating these stakeholders more like customers in the normal world—ensuring smooth study logistics, building easy-to-use data collection tools, and leveraging decentralized trial techniques. Such an approach can enhance participant engagement, reduce dropout rates, and ensure more representative and robust data collection. All of these expedite trial cadence and cut costs, sometimes dramatically. Where enrichment strategies hit their statistical limits, or concerns about external validity or label breadth limit recourse to enrichment, a focus on trial efficiencies may be essential to affordability.
Fortunately, COVID-19 sparked a significant culture shift in how these tools are viewed by important stakeholders in the clinical trial process. Many essential tools for lower-cost, decentralized trials—including video completion of informed consent, remote collection of patient-reported outcomes data, and virtualized clinical visits—are more now widely accepted by regulators and clinicians than before the pandemic.
While these efficiencies can improve productive for nearly all disease areas, they hold particular promise for preventive drugs. As noted above, these products already have higher standards for safety and tolerability, which makes them a natural fit for decentralized trial techniques given the reliance on less direct means of patient supervision. Further, because distributed trials rely less heavily on in-person site visits, they expand the number of eligible and willing patients. This is good for enrollment velocity (and therefore, ultimately, trial affordability), but it can also improve study diversity, which in turn improves the external validity of study results. Trial diversity is also an important issue for the FDA and other regulators for reasons of equity and statistical validity, so this represents another important way to improve study affordability while simultaneously improving data quality.
Conclusions
Preventive anti-infective biologics drugs are a significant advance in healthcare, offering a promising solution to the antibiotic-resistance crisis. By shifting the focus from treatment to prevention, they are poised to transform patient care and the economics of healthcare. The challenges in market dynamics, regulatory considerations, and clinical trial design are substantial, yet surmountable with the straightforward application of a few simple techniques.
References
- Finrow B. 2023. Preventive drugs may be the best solution to the antibiotic resistance crisis. STAT News. https://www.statnews.com/2023/10/18/antibiotic-resistance-crisis-preventive-drugs
- Jester BW, Zhao H, Gewe M, Adame T, Perruzza L, Bolick DT, Agosti J, et al. 2022. Development of spirulina for the manufacture and oral delivery of protein therapeutics. Nature Biotechnology 40(6):956–64.
- Mosbergen D. 2023. The World Needs New Antibiotics, but the Business Model Is Broken. The Wall Street Journal. https://www.wsj.com/tech/biotech/antibiotics-drug-development-business-fda-aa5b4f00
- Lucado J, Gould C, Elixhauser A. 2012. Clostridium difficile infections (CDI) in hospital stays, 2009. Agency for Healthcare Research and Quality (U.S.); Rockville, Md.
- Deshpande A, Pasupuleti V, Thota P, Pant C, Rolston DDK, Sferra TJ, Hernandez AV, Donskey CJ. 2013. Community-associated Clostridium difficile infection and antibiotics: a meta-analysis. Journal of Antimicrobial Chemotherapy 68(9):1951–61.
- McGovern BH, Ford CB, Henn MR, Pardi DS, Khanna S, Hohmann EL, O’Brien EJ, et al. 2021. SER-109, an investigational microbiome drug to reduce recurrence after Clostridioides difficile infection: lessons learned from a phase 2 trial. Clinical Infectious Diseases 72(12):2132–40.
- U.S. Food and Drug Administration. https://www.fda.gov/vaccines-blood-biologics/vowst

Brian Finrow, JD, is Founder and CEO of Lumen Bioscience, a clinical-stage biotechnology company in Seattle, Wash.


