
Clinical Researcher—February 2026 (Volume 40, Issue 1)
PEER REVIEWED
Alexander Kuhn, MS, ACRP-CP; Sophia Charuhas, MA; Catherine Fix, MS; Stacy Kopka, MS; John Tierney, BSN, MPM; Tracey Miller, BSN, RN
The U.S. Food and Drug Administration (FDA) and Office for Human Research Protections (OHRP) have made clear that improving clinical research informed consent is a priority. This article touches on the foundations of informed consent in clinical research and the challenges facing effective informed consent of research participants. In it, the authors explore how clinical research professionals can alter their approach to developing informed consent documents and use both existing resources and incremental change to better serve research participants. They also describe how a small team developed a toolkit composed of visual aids, study visit schedule templates, risk-communication graphics, language repositories, and supplementary guidance documents with the goal of improving informed consent documents and participant understanding. Toolkit development improved awareness of issues with consent form comprehension, and toolkit utilization led to improved informed consent document readability.
Background
Voluntary participation and participant understanding are core to effective informed consent, as established by the Belmont Report, the Nuremberg Code, and the Declaration of Helsinki. However, many consent documents remain difficult for participants to understand in practice, and the effectiveness of informed consent documents continues to be questioned.{1,2}
The FDA has proposed regulatory changes for consistency with the revised Common Rule and has released draft guidance that attempts to provide actionable suggestions for improving the structure of consent forms.{3} Unfortunately, the suggestions leave many details open to interpretation and the scope is primarily limited to the key information section.
Many different explanations exist as to why consent forms fail to effectively inform research participants: they are too long, they are difficult to read and contain extensive sections of legalese, or they confuse participants about the study’s intent, risks, and/or benefits. Likewise, many different strategies have been shown to improve participant understanding: multimedia approaches like PowerPoint presentations or videos, simpler and shorter sentences, adjusting document layout and text styling, or adding pictures.{2,4,5}
While any combination of the suggested causes of poor informed consent or proposed fixes could be the answer, consensus has not been reached,{1,6} and more research is needed to evaluate both the problems and solutions. Practical limitations and situational priorities have a significant impact on what strategies can be implemented. For example, clinical research participants consistently show low levels of comprehension after reading “traditional” informed consents, and e-consenting platforms are proposed as a solution. However, the cost and complexity of implementing these platforms may be prohibitive for many institutions.{7}
This article explores some current thinking on effective informed consent, potential limitations, and how a small group of clinical research professionals leveraged their combined strengths to create an improved consent development methodology that worked within those limitations.
Developing a Strategy
At one time or another, all roles in all phases of research experience pressure to do more with less. The authors of this article comprise a team that is contracted to support the Office of Clinical Research Policy and Regulatory Operations within the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health (NIH). The team assists investigators to more efficiently and effectively develop and navigate initial and amendment approvals of their domestic and international clinical research protocols, consents, and other ancillary documents.
In addition, the team collaborates with colleagues in clinical safety, protocol monitoring, and regulatory affairs offices within NIAID, as well as the NIH Institutional Review Board (IRB) and various other offices within NIH. The goal is to ensure projects align with policies and processes within NIAID, the NIH, or regulatorily, while paying rigorous attention to lessons learned and applying this insight to current projects.
The FDA’s draft guidance and the team members’ ongoing experience with developing informed consents encouraged taking concrete steps to enhance informed consent documents, but it needed to be done within current budgetary and operational constraints. The primary goals were to:
- Improve consent readability and participant comprehension
- Base strategies in the available literature
- Maximize available resources
- Produce sustainable change
The process began by predicting challenges to implementation and anticipating what preparations could be made to avoid obstacles. This meant seeking ongoing feedback during development and implementation to effectively manage expectations, creating evidence-based processes to provide assurance to users, and incrementally developing and implementing changes to ease the transition from current practices.
In addition to referencing publications by OHRP (based within the U.S. Department of Health and Human Services), the NIH Office of Human Subjects Research Protections, the Clinical Trials Transformation Initiative, and the Multi-Regional Clinical Trials Center, PubMed was used to conduct a literature review to explore potential strategies for improving informed consent forms.
Beginning in mid-2023, approximately 30 potentially relevant articles were chosen and reviewed for strategies that could be feasibly implemented, were supported by sufficiently strong evidence, and were relevant to achieving the primary goals as stated above. Findings were routinely presented to leadership to aid the ongoing search and development plans. After several iterations, the strategies were finalized and grouped into “tools” that could be used together in an informed consent “toolkit.”
Each tool was treated as an individual project with its own timeline for development and implementation. Developing one tool at a time had the practical advantage of reducing strain on resources and maximizing the team’s ability to pivot to other priorities as they arose. Furthermore, it naturally led to a stepwise implementation of each tool and thus gradual change for stakeholders. Each tool’s design priority was determined by considering immediate informed consent needs, development burden of the tool, and priorities as communicated by leadership, investigators, and the IRB.
Putting Tools into the Toolkit
Picture Library
The development of the toolkit began with the creation of a comprehensive “picture library”—a curated collection of photos, cartoons, and medical illustrations designed to facilitate communication of information about common procedures found in research consent forms. These images were sourced through targeted web searches, with a focus on identifying resources with unrestricted or broad reuse rights, for example those in the public domain or with Creative Commons licenses. The picture library became the foundation of the toolkit, enabling consent developers to draw from a vetted pool of more than 200 images, rather than conducting time-consuming searches for new images each time they draft a consent.

(An example page from the picture library, including searchable terms and other tracking information.)
Study Visit Schedule Templates
Schedule of events or schedule of activities tables are frequently used to convey study timelines to participants. Many different styles of graphical presentations of study schedules are present in the literature. Those that were determined to be potentially beneficial were refined and compiled into a library of approximately a dozen schemas and graphic schedules of activities templates. Templates are generalized examples that toolkit users can refer to as a starting point to develop a version specific to a particular consent.
Microsoft PowerPoint was chosen to house the template libraries to reduce future design burden by allowing for reuse of similar underlying designs, with the flexibility to tailor each one to a specific study depending on its design, procedures, or focus. IRB-approved versions of templates are placed back into the library for future use and to capture the ongoing preferences of stakeholders.
Icon Library
As the toolkit was incorporated into the consent development process, it became clear that despite an extensive search, the picture library lacked quality entries for some common procedures. The stepwise development of the toolkit allowed for the flexibility to add a new resource to address this issue in real time, and a collection of approximately 250 black and white icons was created, modeled after the picture library.
These simpler icons had the benefit of being small enough to be easily inserted into schemas or study schedules as a complement to, or in place of, text. Icons and pictures both allow for dense sections of text to be broken up or expressed in different ways, with the goal of producing a document that is easier to read and facilitating participant understanding.
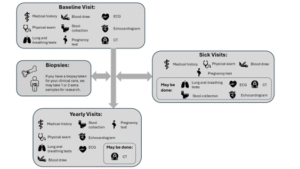
(An example figure developed using a schema template and icons. The same icons used in the schema are then placed elsewhere in the consent form, such as when describing procedures or risks.)
Visually Communicating Risk
Describing what will happen during participation in a study is not limited to an explanation of what the procedures are and when they happen. Informing participants of the risks associated with participation is also an essential part of informed consent. Use of visual aids to enhance participant understanding of risk is not a common best practice for many institutions, despite extensive research demonstrating that the use of visual aids improves document readability and participant understanding of potential risks and benefits of participating in research.{5–7}
To improve participant conceptualization of risk, the toolkit was further expanded to include a small number of templates that visually convey risk to potential participants. Templates included protocol risk summary tables, shaded scales of risk relative to non-protocol risks (e.g., risk of driving a car), body maps to illustrate the location of potential side effects, and arrays of figures with color/shading used to indicate the number of affected persons within a population to aid in understanding the likelihood of experiencing a given risk.
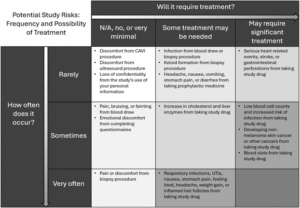
(An example risk summary table, envisioned as a way for a participant to have study risks contextualized by frequency and treatment.)
Supplementary Materials
In conjunction with these libraries, supplementary materials were created to aid in the consent development process, including:
- A best practices document. This document created a centralized source for disparate research findings and reviewer feedback on best practices for informed consent writing. Consolidating these guidelines into a centralized location streamlined the consent development workflow, and improved efficiency and consistency across consents.
- A repository of risk language organized by procedure and date/circumstances of IRB/ethics committee (EC) approval. This facilitated effective and efficient choice of language for several dozen procedures that have been acceptable to subject matter experts (investigators and medical writers) and the IRB/EC.
- Instructions and guidelines for the use of a program called Visual Basic. Visual Basic is part of Microsoft Word and allows users to create new automated editing functionalities. This augments the program’s ability to automatically suggest word choice, active voice, and stylistic improvements with the goal of improving document readability and participant comprehension.
- Guidance document for implementation of artificial intelligence (AI) resources. Though not available when the toolkit began development, AI image generation has become more widely available and can be particularly helpful in customizing images to a target population. Large language models are also garnering increasing interest, though the ethical and appropriate application of both these modalities is still evolving. This document is updated as new information becomes available, and other resources within the toolkit receive related updates, as applicable.
Implementing Your Strategy and Lessons Learned
Keeping a distinct methodology in mind will reduce the burden of choosing which tools to use for a particular consent form. Each tool or template has its own benefits and drawbacks, and will not be suitable for all situations. Allowing authors to choose when a tool does or does not benefit the participant and the study empowers users and engenders ownership of the tools and methodology.
When designing tools, build them in a way that makes it clear to an inexperienced user how the tool is intended to be used. However, even with guidance, people may not use the tools as intended; this can be an opportunity. The tools are not intended to be static and unchanging, as new evidence becomes available, the tools should be updated. This can either be from a concerted effort to update all the tools, for example in response to a change in methodology, or simply from user feedback.
Embrace the knowledge and creativity of people using the tools and encourage feedback. This allows for iterative improvement of the tools—to either incorporate new ideas and uses, or to update instructional language to clarify issues for users. Toolkit development can also be incorporated into an ongoing process, such as a standing team meeting. Team members discuss successes, failures, and opportunities to advance a particular tool or workflow, and this keeps the resources current and flexible.
Consent development depends on a variety of factors. When developing and implementing a strategy to change how you prepare and use informed consent forms, some questions that deserve attention are:
- Who is your intended audience?
- If amending a currently approved consent, should toolkit resources be incorporated?
- What will encourage use of the tools?
- How can stakeholder expectations be managed?
- Will stakeholder feedback be incorporated and, if so, when and how?
- Which tools do you think will most improve the participant/reader experience?
- Will research staff need training on changes from their “typical” processes?
- What financial or logistical limitations can you foresee?
Clearly, more research is needed to determine what methodologies are best for each situation, and significant resource constraints are a reality for many researchers. While new research should always be welcomed and encouraged, and challenges to implementation will persist, creating necessary change cannot always wait.
In the authors’ case, the toolkit has contributed to a reduced document reading level and consents utilizing the toolkit resources have received praise from investigators and IRB reviewers. Lasting change comes from an accumulation of small changes, and making the most of the resources already on hand organically creates incremental improvement.
Start a dialogue with IRB/EC colleagues about how to address deficiencies. Talk with investigators about incorporating a new technique into their consenting process. Knowledge of the issues and drive to see a positive change for both the clinical research enterprise and the clinical research participant are critical resources—do not let them go to waste.
Special thanks to Ben Snow and Kristin Young for their contributions to the development of the toolkit.
Funding Statement
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. 75N910D00024. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
References
- Grant SC. 2021. Informed Consent—We Can and Should Do Better. JAMA Network Open4(4):e2110848. https://doi.org/10.1001/jamanetworkopen.2021.10848
- Wisgalla A, Hasford J. 2022. Four reasons why too many informed consents to clinical research are invalid: A critical analysis of current practices. BMJ Open12(3):e050543. https://doi.org/10.1136/bmjopen-2021-050543
- U.S. Food and Drug Administration. 2024. Key Information and Facilitating Understanding in Informed Consent – Guidance for Sponsors, Investigators, and Institutional Review Boards: Draft Guidance. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/key-information-and-facilitating-understanding-informed-consent-guidance-sponsors-investigators-and
- Flory J, Emanuel E. 2004. Interventions to improve research participants’ understanding in informed consent for research: A systematic review. JAMA292(13):1593–1601. https://doi.org/10.1001/jama.292.13.1593
- Nishimura A, Carey J, Erwin PJ, Tilburt JC, Murad MH, McCormick JB. 2013. Improving understanding in the research informed consent process: A systematic review of 54 interventions tested in randomized control trials. BMC Medical Ethics14:28. https://doi.org/10.1186/1472-6939-14-28
- Coyle M, Gillies K. 2020. A systematic review of risk communication in clinical trials: How does it influence decisions to participate and what are the best methods to improve understanding in a trial context? PloS One15(11):e0242239. https://doi.org/10.1371/journal.pone.0242239
- Lentz J, Kennett M, Perlmutter J, Forrest A. 2016. Paving the way to a more effective informed consent process: Recommendations from the Clinical Trials Transformation Initiative. Contemporary Clinical Trials49:65–9. https://doi.org/10.1016/j.cct.2016.06.005
Alexander Kuhn, MS, ACRP-CP, (alexander.kuhn@nih.gov) is a Protocol Navigator in the Clinical Monitoring Research Program Directorate with the Frederick National Laboratory for Cancer Research.
Sophia Charuhas, MA, is a Medical Writer in the Clinical Monitoring Research Program Directorate with the Frederick National Laboratory for Cancer Research.
Catherine Fix, MS, is a Medical Writer in the Clinical Monitoring Research Program Directorate with the Frederick National Laboratory for Cancer Research.
Stacy Kopka, MS, is Medical Writer Manager in the Clinical Monitoring Research Program Directorate with the Frederick National Laboratory for Cancer Research.
John Tierney, BSN, MPM, is in the Office of Clinical Research Policy as Regulatory Operations Director with the National Institute of Allergy and Infectious Diseases, part of the National Institutes of Health.
Tracey Miller, BSN, RN, is Protocol Navigation Director in the Clinical Monitoring Research Program Directorate with the Frederick National Laboratory for Cancer Research.


